


Guidance for Industry
Manufacturing Biological Intermediates and Biological Drug Substances Using Spore-Forming Microorganisms
[PDF printable version - 185 KB]
Additional copies of this guidance are available from the Office of Communication, Training and Manufacturers Assistance (HFM-40), 1401 Rockville Pike, Suite 200N, Rockville, MD 20852-1448, or by calling 1-800-835-4709 or 301-827-1800, or from the Internet at http://www.fda.gov/cber/guidelines.htm.
For questions on the content of this guidance, contact Cynthia Kelley at 301-827-0636 or John Eltermann at 301-827-3031.
U.S. Department of Health and Human Services
Food and Drug Administration
Center for Biologics Evaluation and Research
September 2007
- INTRODUCTION
- SCOPE
- BACKGROUND
- MANUFACTURING WITH SPORE-FORMERS IN A SEPARATE DEDICATED BUILDING
- Physical Establishment and Equipment
- Containment
- Building Construction and Configuration
- Air Handling Units (AHUs)
- Equipment Dedication
- Procedural Control
- Personnel Gowning and Flows
- Material Transfer
- Equipment Cleaning
- Waste Disposal
- MANUFACTURING WITH SPORE-FORMERS IN A MULTIPRODUCT MANUFACTURING BUILDING: PROCESS CONTAINMENT
- Physical Establishment and Equipment
- Containment
- Building Construction and Configuration
- Air Handling Units (AHUs)
- Equipment Dedication
- Procedural Control
- Personnel Gowning and Flows
- Material Transfer
- Equipment Cleaning
- Waste Disposal
- Campaign Changeovers
- Monitoring Specific for the Spore-Former
- Environmental Monitoring Specific for the Spore-Former
- Test Methods Specific to the Spore-Former
- Specificity
- Sample Recovery
- Detection Limit
- Sample Locations
- SPILL CONTAINMENT
- MAINTENANCE AND DECOMMISSIONING
- Maintenance
- Decommissioning
- DEFINITIONS
APPENDIX: SPORE-FORMING MANUFACTURING DIAGRAM
Contains Nonbinding Recommendations
Guidance for Industry
Manufacturing Biological Intermediates and Biological Drug Substances Using Spore-Forming Microorganisms
| This guidance represents the Food and Drug Administration's (FDA's) current thinking on this topic. It does not create or confer any rights for or on any person and does not operate to bind FDA or the public. You can use an alternative approach if the approach satisfies the requirements of the applicable statutes and regulations. If you want to discuss an alternative approach, contact the appropriate FDA staff. If you cannot identify the appropriate FDA staff, call the appropriate number listed on the title page of this guidance. |
- INTRODUCTION
- SCOPE
- BACKGROUND
- MANUFACTURING WITH SPORE-FORMERS IN A SEPARATE DEDICATED BUILDING
- Physical Establishment and Equipment
- Containment
- Building Construction and Configuration
- Air Handling Units (AHUs)
- Equipment Dedication
- Procedural Control
- Personnel Gowning and Flows
- Material Transfer
- Equipment Cleaning
- Waste Disposal
- MANUFACTURING WITH SPORE-FORMERS IN A MULTIPRODUCT MANUFACTURING BUILDING : PROCESS CONTAINMENT
- Physical Establishment and Equipment
- Containment
- Building Construction and Configuration
- Air Handling Units (AHUs)
- Equipment Dedication
- Procedural Control
- Personnel Gowning and Flows
- Material Transfer
- Equipment Cleaning
- efficacy of the decontamination;
- analysis of potential residue for the specific spore-former;
- evaluation of product contact surface areas, and the interior and exterior areas on equipment to include controls, valves, seals, probes, motors, wiring harnesses, and miscellaneous external surfaces;
- sampling procedures; and
- analytical methods, including sensitivity of those methods.
- Waste Disposal
- Campaign Changeovers
- Monitoring Specific for the Spore-Former
- Environmental Monitoring Specific for the Spore-Former
- Test Methods Specific to the Spore-Former
- Specificity
- Sample Recovery
- Detection Limit
- Sample Locations
- SPILL CONTAINMENT
- MAINTENANCE AND DECOMMISSIONING
- Maintenance
- Decommissioning
- DEFINITIONS
The former regulations at 21 CFR 600.11(e)(3) (§ 600.11(e)(3)) required that all work with spore-bearing microorganisms (spore-formers) be conducted in an entirely separate building, or in a completely walled-off portion of a multiproduct building. This isolated building had to be dedicated exclusively for the manufacturing or storage of spore-formers. Previously, areas in a multiproduct building used for manufacturing with spore-formers were required to be constructed to prevent cross-contamination of other areas, and were required to include entrances that were separate and independent from the remainder of the facility. All equipment used for manufacturing spore-formers was to be permanently identified and reserved exclusively for use with those microorganisms. Any materials destined for further manufacture were to be removed from this area only under conditions that prevented the introduction of spores into other manufacturing areas.
We, the Food and Drug Administration (FDA), modified the regulatory requirements for the manufacturing of biological products with spore-formers to allow greater manufacturing flexibility. Under the revised regulation1 we no longer require the use of permanently dedicated buildings and equipment for spore-formers, if certain controls and precautions are applied. We recognize that advances in facility, system, equipment design, testing, and sterilization technologies have increased the ability of manufacturers to control and analyze the manufacture of biological products. As industry has gained experience with these new technologies, we found that manufacturers could evaluate aspects of a biological product's safety and purity with testing. The use of appropriate procedural controls, validated processes, and enhanced testing capability provide the manufacturer with a degree of confidence that their biological product achieves the expected levels of safety and purity. Areas of special concern, such as process containment, contamination with pathogenic and/or toxic agents, sterilization, and disinfection can be addressed using currently available procedures and processes.
We recognize that spore-formers are currently used in manufacturing processes and there will likely be a need to use them in the immediate future. However, for the production of future biological products, manufacturers are encouraged to identify alternatives to the use of spore-forming microorganisms whenever possible. Such alternatives could include the use of sporulation deficient strains, or recombinant proteins expressed in nonspore-forming microorganisms. We anticipate that certain second-generation vaccines, including those against anthrax, botulism, and tetanus will likely contain recombinant proteins. The revised regulation uses the term "spore-forming" microorganisms to describe organisms that are capable of spore production. The term "spore-bearing" microorganism, which was used in the earlier regulation, is not used because the term spore-forming microorganism is the more commonly accepted description of this class of microorganisms. For the purposes of this guidance, the term spore-forming microorganism (or "spore-former") includes both the spore and vegetative forms of the organism.
This guidance document finalizes the draft guidance entitled "Guidance for Industry: Manufacturing Biological Drug Substances, Intermediates, or Products Using Spore-Forming Microorganisms " dated February 2005 ( February 24, 2005; 70 FR 9084).
FDA's guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidance describes the agency's current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in agency guidance means that something is suggested or recommended, but not required.
The purpose of this document is to provide to you, manufacturers of biological intermediates and biological drug substances using spore-forming microorganisms, guidance in response to changes made to § 600.11(e)(3). The revised regulation describes the requirements for manufacturing using spore-forming microorganisms and allows manufacturers greater flexibility than under the prior regulation. We are limiting the scope of this guidance to biological intermediates and biological drug substances as spore-formers should not be present in the finished dosage form (biological drug product). Once the biological intermediates or biological drug substances have been separated from the viable spore-forming microorganism, the concern for cross-contamination diminishes. We recommend that you establish a distinct crossover point in the separation process.
This guidance applies to biological manufacturing processes utilizing spore-forming microorganisms regulated under the Federal Food, Drug and Cosmetic Act (the Act), the Public Health Service Act (PHS Act) and the applicable regulations in 21 CFR Parts 600 through 680. Section 501(a)(2)(b) of the Act requires that the methods used in, or the facilities or controls used for, the manufacture, processing, packing or holding of drugs, including biological intermediates and biological drug substances, conform to or are operated and administered in conformity with current good manufacturing practice to assure that such drug meets the requirements of the Act as to safety and has the identity and strength, and meets the quality and purity characteristics, which it purports or is represented to possess.
This guidance does not apply to allergenic and fungi source material, in vitro diagnostics, or therapeutics. Additionally, this guidance does not apply to spore-forming microorganisms used for the supplemental sterilization procedure control test described in § 600.11(e)(2).
When the spore-former used in the manufacturing process is a pathogen or is potentially pathogenic, the stricter requirements related to equipment and supplies between § 600.11(e)(5) and revised § 600.11(e)(3) must be applied.
Spore-formers are used in the production of certain biological products. Biological intermediates or biological drug substances derived from spore-formers may be used as source material for further manufacture into final biological drug products, such as vaccines. Bacteria produce spores as a means to survive adverse environmental conditions. In general, spores show enhanced resistance over bacteria to high temperatures, freezing, dryness, antibacterial agents, radiation, and toxic chemicals. Under favorable conditions, spores can germinate into actively growing vegetative bacterial cells. Some of these spore-formers are human pathogens and are associated with high morbidity and mortality. Due to their unique survival properties, spore-formers pose great challenges to manufacturers. In order to ensure the safety of a product manufactured in a facility in which spore-formers are present, these microorganisms must be kept under stringent control in order to avoid the release of spores into the manufacturing area where they have the potential to cross-contaminate other products and areas. (§ 600.11(e)(3)(i)).
Manufacturing with spore-formers requires varying levels of control depending on the characteristics (e.g., virulence toward humans) of the microorganism that is utilized in a manufacturing process. The level of confidence a specific process provides for safety and purity may also be a factor in deciding the level of manufacturing control necessary. You must institute the appropriate controls in the manufacturing facility, associated equipment, and manufacturing procedures to avoid contamination. (§§ 600.11(a), 600.11(e)(3)(i), 600.11(e)(5)). The recommendations described in this guidance are intended for manufacturing with the type of spore-formers that are currently used by the regulated industry and included in the scope of this guidance. Any atypical or novel spore-formers may require specific controls that are unique for that particular microorganism.
Prevention of spore contamination can be achieved by using a separate dedicated building or by using process containment if manufacturing is conducted in a multiproduct manufacturing building. Containment is established by facility design and/or the use of procedures and equipment that prevent the release of spores into adjacent areas. (§ 600.11(e)(3)).
This section applies if you choose to use a separate dedicated building configuration for manufacturing with spore-formers. Although you are no longer required to use this type of configuration, if you do so, you must follow the applicable requirements in § 600.11(e)(3)(i) to avoid having to satisfy the process containment requirements in subsection (e)(3)(ii). This section of the guidance document reiterates the requirements in subsection (e)(3)(i) applicable to separate dedicated buildings, and provides additional recommendations for using this configuration.
The use of a separate dedicated building for manufacturing processes involving spore-formers is the simplest means to prevent cross-contamination. The separate dedicated building configuration is not intended to accommodate the manufacturing of multiple products using spore-formers. If multiple products are manufactured in the same area or within the same building using spore-formers, then additional criteria will apply. See § 600.11(e)(3)(ii) and section V. below.
If you choose to use a separate dedicated building configuration for manufacturing processes involving spore-formers, your building must be constructed to prevent contamination. (§ 600.11(e)(3)(i)). Your separate dedicated building should be constructed with walls that extend to the ceiling with properly sealed joints at wall/ceiling intersections, separate heating, ventilation, and air conditioning (HVAC), water drops, sewer line, and an independent entrance thereby containing the building from surrounding areas.
We recommend that all surfaces be solid, hard, non-porous, and cleanable, including ceilings and walls. To the extent you are using aseptic processing, floors, walls, and ceilings should have smooth, hard surfaces that are easily cleanable.2
AHUs should not be shared with other buildings. Dedicated re-circulating AHUs are acceptable within the separate dedicated building. We recommend that exhaust not be located near other AHU intakes. We also recommend that you consider using high efficiency particulate air (HEPA) filtration of exhaust, and that you maintain the building at negative pressure with respect to the outside environment and/or adjacent areas.
If you choose a separate dedicated building configuration, major equipment should be identified as used for manufacturing with the particular spore-former. We recommend that you dedicate all equipment.
Personnel who work in manufacturing using spore-formers must complete an outer covering change or wear protective covering prior to entering areas where other products are manufactured (§ 600.10(c)(3)). We also recommend that personnel shower and complete a "clean" clothing change prior to entering other areas or interacting with personnel not directly involved in the manufacturing of spore-formers.
Any material transferred out of a spore-former manufacturing area that may cross-contaminate other products, product containers, intermediates, or materials used in the manufacturing of other products should be decontaminated via a decontamination chamber prior to exiting. If a decontamination chamber is not feasible, then a series of airlocks utilizing surface decontamination with an appropriate agent may be acceptable.
When utilizing a decontamination chamber system such as an autoclave, sterilize-in-place (SIP) cycle, or gas chamber, the decontamination cycle should be validated using methodologies that ensure that the spore-former would be inactivated. We recommend that you incorporate a worst-case approach. You should not use the same chamber for both the decontamination of the spore-former and the sterilization of other production items.
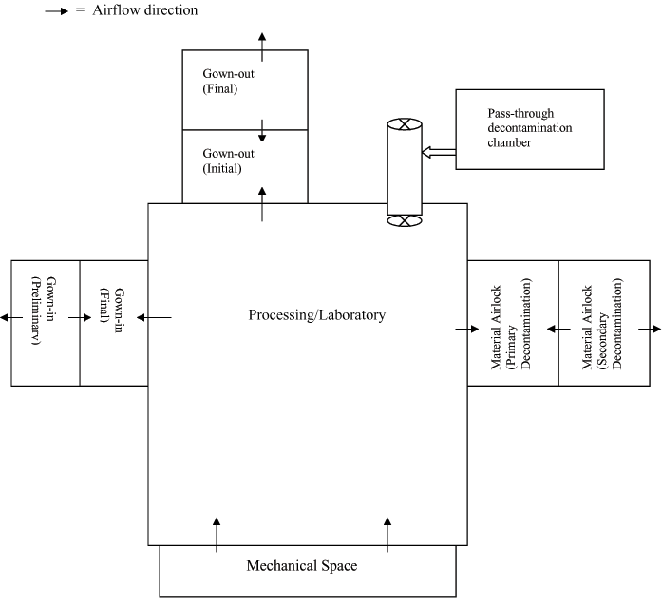
When it is necessary to employ an airlock system for decontamination of material that is transferred out of a manufacturing area, your procedures should provide for the decontamination process. Use of a liquid or gaseous agent to inactivate the spore-formers and decontamination agent efficacy studies are recommended. We recommend that you incorporate a worst-case approach. We also recommend that you consider parameters that include the composition of the material, exposure time, concentration, pH, temperature and any other relevant factors. If the airlock system involves only local decontamination of the equipment or item, such as wipe downs, then we recommend that the decontamination process be conducted and validated in series (multiple airlocks) to ensure that no trailing contamination is encountered. We recommend that inside personnel pass the outgoing material and/or equipment into an interior material airlock (i.e., Material Airlock (Primary Decontamination) as shown in the Appendix), where they decontaminate the items according to approved validated procedures. After initial decontamination, the inside personnel transfer the outgoing items into an exterior material airlock (i.e., Material Airlock (Secondary Decontamination) as shown in the Appendix). The outside personnel then provide a second decontamination step (also validated) on the outgoing items, and then transfer the material and/or equipment into an exit corridor or room. We recommend that temporal separation between personnel be maintained and that personnel not use material airlocks for exiting (see Appendix).
No special equipment cleaning is needed, beyond what § 600.11(b) requires, when manufacturing with spore-formers, provided that the equipment is maintained within the dedicated processing area.
Process waste can be treated in a similar fashion as the material transferred out of the manufacturing area or processed through a validated bio-waste system. When waste is transferred out of the manufacturing area, we recommend that it be transferred out in biohazard bags via multiple airlocks in a manner similar to the material transfer process described in section IV.A.2.b. We recommend that waste be bagged in the processing room and transferred to the interior material airlock where the outside of the bag is decontaminated by the inside personnel. After the initial decontamination, inside personnel places the bagged waste into another biohazard bag and transfers it into the exterior material airlock. Outside personnel decontaminates the outside of the second biohazard bag and removes the double-bagged waste from the exterior material airlock (see Appendix).
All waste must be disposed of in a manner that is in compliance with federal, state, and local environmental laws.
Process containment is designed to isolate equipment or an area that involves manufacturing using spore-forming microorganisms mechanically (Containment) and procedurally (Procedural Control). This manufacturing area, including biological intermediates or biological drug substances, equipment, or material storage in that area, must not be used for any other purpose during the processing period (§ 600.11(e)(3)(ii)). The manufacturing area should contain adequate space for the orderly placement of equipment. If you intend to use an area for manufacturing spore-formers and other products on a campaign basis, then you must develop and implement procedures to demonstrate containment and decontamination before initiating processing of other products in that manufacturing area (§ 600.11(e)(3)(ii)). These procedures must, at a minimum, provide for containment, environmental monitoring specific for the spore-former, cleaning, decontamination, and movement of materials in and out of that area (§ 600.11(e)(3)(ii)). We recommend that you implement campaign changeover procedures whenever an area that was previously used for a spore-forming microorganism will subsequently be used for manufacturing other products.
We recommend that the manufacturing areas used for spore-forming microorganisms have double serial entry, exit, and material airlocks to minimize the potential for cross-contamination via personnel, materials, and air turbulence. As shown in the Appendix, the interior airlocks would serve as an air sink for the primary decontamination step. The three air sinks shown in the Appendix include the final gowning step (Gown-in (Final)), the gross de-gowning step (Gown-out (Initial)), and the interior material airlock (Material Airlock (Primary Decontamination)). The exterior airlocks would serve as an air dome for the preliminary gowning step. The air domes shown in the Appendix include primary decontamination (Gown-in (Preliminary)), the final de-gowning step (Gown-out (Final)), and the secondary decontamination step (Material Airlock (Secondary Decontamination)).
We recommend that all surfaces be solid, hard, non-porous, and cleanable, including ceilings and walls. To the extent you are using aseptic processing, floors, walls, and ceilings should have smooth, hard surfaces that are easily cleanable. We recommend that the final ceiling and wall finishes minimize air leaks.
Your procedures should provide for the decontamination of all tables, shelving, and storage apparatus.
We recommend that AHUs using 100% single-pass air be used within the area where manufacturing with spore-forming microorganisms occurs and that the area be maintained at a negative air pressure to all surrounding areas, including ceilings and mechanical spaces. We recommend that exhaust not be located near other AHU intakes, and that HEPA filtration of exhaust be considered.
Wherever possible, we recommend that major processing equipment be dedicated for a specific product use and that such equipment be identified to show the specific equipment used in the manufacture of each batch of product. If equipment is dedicated for a specific manufacturing process using a spore-former, it must be decontaminated prior to the introduction of a new production process (§ 600.11(e)(3)(ii)), and we recommend removing it from the area, where feasible. We recommend that dedicated equipment be segregated from other equipment while in storage.
We recommend that small ancillary equipment and administrative items such as pens, logbooks, pipettes, miscellaneous glassware, and standard operating procedures (SOPs) be dedicated or made disposable. Dedicated and disposable items must be decontaminated and/or removed from the processing area during the area changeover procedure (§ 600.11(e)(3)(ii)).
When equipment dedication and removal is not feasible, such as with large fixed mounted bioreactors, decontamination and cleaning procedures must be in place (§ 600.11(e)(3)(ii)). See also section V.A.2.c. below.
We recommend that personnel entry and exit flows be unidirectional. We also recommend that personnel gown with multiple layers so that personnel gowning and de-gowning occurs in multiple stages (see Appendix) and that gowning procedures are designed to prevent or reduce the potential for personnel to carry the spore-former out of the area via their hair, skin, shoes, jewelry, or clothing. We recommend that disposable gowning be decontaminated using a validated process or adequately bagged and sealed prior to leaving the area used for spore-forming microorganisms. Personnel who work in manufacturing using spore-formers must complete an outer covering change or wear protective covering prior to entering areas where other products are manufactured (§ 600.10(c)(3)). We also recommend that personnel shower and complete a "clean" clothing change prior to entering other areas or interacting with personnel not directly involved in the manufacturing of spore-formers.
The recommendations for material transfer in a multiproduct manufacturing building are the same as those for a separate dedicated building (see section IV.A.2.b.).
When the use of dedicated or disposable equipment is not feasible, such as with large or fixed bioreactors, decontamination and cleaning procedures must be in place (§ 600.11(e)(3)(ii)). In development of these procedures for each piece of equipment you should consider the following:
We recommend the dismantling and inspection of such equipment between different products. Some cleaning agents (e.g., NaOH) can lead to pitting of equipment and may protect spore-formers from the decontaminant.
The recommendations for waste disposal in a multiproduct manufacturing building are the same as those for a separate dedicated building (see section IV.B.).
Campaign changeovers involve the cleaning and decontamination of a specific area that has been exposed to a spore-former in preparation for the introduction of another product or process into that same area. The decontamination and cleaning would include all equipment and items that may have been exposed to the spore-forming microorganism. This includes Biological Safety Cabinets and re-circulating AHUs. In addition, consideration may be given to decontamination of the entire manufacturing area with a gaseous sterilant such as chlorine dioxide or hydrogen peroxide.
Prior to introducing a new product into a facility previously used to manufacture spore-formers, you must develop and implement procedures that focus on issues related to containment, environmental monitoring specific for the spore-former, cleaning, decontamination, and movement of equipment and materials in and out of that area (§ 600.11(e)(3)(ii)). Campaign changeovers as outlined in this document occur when an area that was previously used for spore-former manufacturing will subsequently be used for manufacturing any other product.
We recommend that the following steps be taken in the order indicated below for cleaning and decontamination of an area that is to be used on a campaign changeover:
1) All waste in the area is removed or sent to the bio-waste system, as required by § 600.11(e)(3)(ii);
2) Stay-in-place equipment is decontaminated, as required by § 600.11(e)(3)(ii);
3) Removable dedicated equipment and ancillary items are decontaminated and removed or sent to waste for disposal from the area according to material transfer procedures, as required by § 600.11(e)(3)(ii) and as outlined with additional specific recommendations in section IV.A.2.b.;
4) Room and room fixtures (e.g., shelves, incubators, storage units) are decontaminated and cleaned according to procedures, as required by § 600.11(e)(3)(ii);
5) Stay-in-place processing equipment is dismantled, cleaned, and sterilized (if applicable);
6) Environmental monitoring specific for the spore-formers used in manufacturing is performed, as required by § 600.11(e)(3)(ii) (see section V.D. for additional recommendations); and
7) A quality control unit review of the campaign changeover data (including environmental monitoring results) and a quality control unit area inspection are executed prior to releasing the area for the next product.
Environmental monitoring3 that is specific for the spore-former must be conducted in adjacent areas during manufacturing operations and in the manufacturing area after completion of cleaning and decontamination, at the conclusion of the campaign changeover procedures (§ 600.11(e)(3)(ii)). During operations, we suggest that the sampling and testing be conducted in the adjacent areas at the beginning, middle, and end of each manufacturing shift. All areas must be free from contamination with the spore-former. (§ 600.11(e)(3)(i)).
Test methods used for monitoring specific for the spore-former should be qualified for specificity, sample recovery, and detection limits, at a minimum. A quantitative or qualitative test is acceptable provided the test can be appropriately qualified.
Testing should be able to detect the specific spore-former and identify it in the presence of other microorganisms.
Sampling qualifications should demonstrate the percent recovery of the specific spore-former.
Limits of detection using the specific spore-former should be established.
We recommend the adjacent areas, associated with the isolated processing area, be sampled near or at the points of possible egression, such as doorways, windows, and/or other openings.
We recommend that the number of samples for testing at the conclusion of the campaign changeover be developed by a matrix approach and take into account the size and complexity of the equipment, room, and room fixtures. We suggest that the exact sampling sites be determined based on equipment and items that are the most difficult to clean and decontaminate and the likelihood of product impact. For example, controls over a formulation tank should be tested, but the floor in an isolated corner may not need to be included.
We recommend that procedures be in place to address emergency responses for all spills involving spore-forming microorganisms regardless of location. These procedures should include validated processes for containing the spill, and cleaning and decontaminating the area and equipment affected by the spill.
We recommend that maintenance activities be performed under appropriate controls to prevent contamination of other products and areas within the site. If it is necessary to break the integrity of the process containment to perform maintenance activities, then you must follow the established decontamination procedures prior to the maintenance activities and after all manufacturing activity has ceased. (§ 600.11(e)(3)(ii)).
Decommissioning of a dedicated spore-former manufacturing area or building must involve a comprehensive decontamination plan with extensive spore-former specific testing to ensure the adequate removal and/or cleaning of the area and building prior to the introduction of another product (see section V.D.). (§ 600.11(e)(3)(ii)).
a. Air handling unit (AHU) - A component of the HVAC system that includes the fans, filters, coils, and other materials used to generate conditioned air.
b. Biological drug product - A finished dosage form that contains an active biological drug ingredient generally, but not necessarily, in association with inactive ingredients.
c. Biological drug substance - Any substance or mixture of substances intended to be used in the manufacture of a biological drug product and that, when used in the production of a biological drug product, becomes an active ingredient of the biological drug product. Such biological substances are applicable to the prevention, treatment, or cure of a disease or condition of human beings.
d. Biological intermediates - A material produced during steps of the processing that must undergo further molecular change or purification before it becomes a biological drug substance. Biological intermediates may or may not be isolated.
e. Biological product - Any virus, therapeutic serum, toxin, antitoxin, vaccine, blood, blood component or derivative, allergenic product, or analogous product, or arsphenamine or derivative of arsphenamine, applicable to the prevention, treatment, or cure of disease or condition of human beings.
f. Campaign changeovers - The process of cleaning and decontaminating a specific area, equipment, system, waste, and/or ancillary items exposed to a spore-forming microorganism in preparation for the introduction of another product or process into that same area.
g. Containment - Physical/mechanical barriers and/or systems designed to control spore-formers to prevent contamination.
h. Cross-contamination - Contamination of a material or product with another material or product.
i. Crossover point - The processing point where viable organisms are not part of the process or process solutions.
j. Decontamination of spore-forming microorganisms - A process that eliminates or inactivates viable bioburden and any associated toxic material via the use of chemical and/or physical means.
k. Facility - Physical structure.
l. Manufacturing area - A specified location within a facility associated with the manufacturing of any one product or multiple products.
m. Manufacturing facility - The physical structure associated with the manufacturing of any one product or multiple products.
n. Manufacturing site - The entire complex of buildings, connected or separate, and belonging to one entity engaged in the manufacturing of any one product or multiple products.
o. Multiproduct - More than one approved product, licensed product, investigational new drug product or separate process.
p. Procedural control - Manufacturing procedures executed in such a manner as to prevent or minimize spore-former contamination.
q. Process containment - A system designed to mechanically and procedurally isolate equipment or an area that involves manufacturing using spore-forming microorganisms.
r. Qualification - Action of proving and documenting that equipment, ancillary systems, or areas are properly designed, installed, work correctly, and actually lead to the expected results. Qualification is part of validation, but the individual qualification steps alone do not constitute process validation.
s. Spore-forming microorganism - Organisms that are capable of spore production, including both spore and vegetative forms of the organism.
t. Validation - Establishing documented evidence that provides a high degree of assurance that a specific process will consistently produce a result meeting its predetermined specifications and quality attributes.
APPENDIX: SPORE-FORMING MANUFACTURING DIAGRAM
Footnotes
1 In the Federal Register of December 30, 2003, FDA published for public comment the Direct Final Rule entitled, "Revision of the Requirements for Spore-Forming Microorganisms" (68 FR 75116), and the accompanying Proposed Rule entitled, "Revision of the Requirements for Spore-Forming Microorganisms; Companion to Direct Final Rule" (68 FR 75179). In the Federal Register of May 14, 2004, FDA published the "Revision of the Requirements for Spore-Forming Microorganisms; Confirmation of Effective Date" (69 FR 26768) confirming the effective date of June 1, 2004, for the direct final rule.
2 Please refer to FDA's guidance for industry entitled "Sterile Drug Products Produced by Aseptic Processing - Current Good Manufacturing Practice," dated September 2004 (October 4, 2004, 69 FR 59258) (http://www.fda.gov/cder/guidance/5882fnl.htm), for the agency's current thinking on aseptic processing.
3 Please refer to FDA's "Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing - Current Good Manufacturing Practice" dated September 2004 (October 4, 2004, 69 FR 59258) (http://www.fda.gov/cber/gdlns/steraseptic.htm), for the agency's current thinking on aseptic processing and environmental monitoring.