Attachment to Guidance on Pharmacogenomic Data Submissions
Examples of Voluntary Submissions or Submissions Required Under 21 CFR 312, 314, or 601
[PDF version]
U.S. Department of Health and Human Services
Food and Drug Administration
Center for Drug Evaluation and Research (CDER)
Center for Biologics Evaluation and Research (CBER)
Center for Devices and Radiological Health (CDRH)
March 2005
Procedural

Contains Nonbinding Recommendations
Pharmacogenomic Data Submissions
Attachment: Examples of Required or Voluntary Submissions
This attachment to the guidance Pharmacogenomic Data Submissions is intended to illustrate when it would be appropriate to submit a voluntary (VGDS) genomic data submission versus a pharmacogenomic data submission required under 21 CFR 312, 314, or 601 (RGDS). Please refer to the complete guidance, or contact the relevant center if you have any questions. Examples for various topic areas are provided using the following format:
Scenario
Type of Submission
Rationale
Topic Area: Metabolizing Enzymes
Scenario 1: During IND development, a sponsor conducts single- and multiple-dose pharmacokinetic studies of a new molecular entity (NME) in healthy volunteers enrolled to represent the major racial demographic groups. The NME is metabolized primarily by CYP2C19 to inactive metabolites. The sponsor assesses the CYP2C19 genotypes in the volunteers to determine the clearance phenotype with the goal of determining if drug dosing needs to be individualized based on the genotype groups.
Type of Submission:
Required in full report (IND)
Rationale:
The sponsor uses the test results to "support scientific arguments pertaining to the pharmacologic mechanism of action, the selection of drug dosing or the safety and effectiveness of a drug" (as described in Figure A2 of this document).
Scenario 2: A sponsor conducts a phase 3 clinical trial of a NME in patients with the target indication. The NME is metabolized primarily by CYP2D6 to an active metabolite equipotent to the parent molecule. The sponsor genotypes a randomly selected subset of the patients for their CYP2D6 alleles to explore the association between genotype, drug dosing, and clinical outcome. The results show minor differences in clinical outcomes among the genotypes. The information is included in the proposed labeling in the NDA submission.
Type of Submission:
Required in full report (NDA)
Rationale:
The sponsor included the test results in the drug label (as described in Figure B1 of this document).
Scenario 3: A sponsor conducts a phase 3 clinical trial of a NME in patients with the target indication. The NME is metabolized primarily by CYP2D6 to an active metabolite equipotent to the parent molecule. After the trial is completed, the sponsor genotypes a randomly selected subset of the patients for their CYP2D6 alleles to explore the association between genotype and clearance values. The sponsor will not include the results in the labeling.
Type of Submission:
Required in abbreviated report (IND or NDA/BLA)
Rationale:
Although the test results were not used in decision-making or scientific arguments (such as described in Figure A1 or A2) or in the drug label or as part of the scientific database (such as described in Figure B1), CYP2D6 is a known valid biomarker. Therefore, the test results must be submitted as an abbreviated report (as described in Figure A3, Figure B2 of this document).
Scenario 4: A sponsor conducts a drug interaction study in healthy volunteers of their NME, a CYP3A substrate, co-administered with ketoconazole as an enzyme inhibitor. Subsequent to the study, the subjects are genotyped for their CYP3A5 alleles to determine the relative contribution of this polymorphism to inter-individual variability in AUC.
Type of Submission:
For submissions under an IND, these data could be submitted as a VGDS. For submissions under NDA/BLA, these data would be required to be submitted as a synopsis, and a VGDS of the data is encouraged.
Rationale:
The test results are not being used in decision-making or scientific arguments (such as described in Figure A1 or A2) or in the drug label or as part of the scientific database (such as described in Figure B1). In addition, polymorphism of CYP3A5 is not widely studied and is therefore neither a probable or known valid biomarker (such as described in Figure A3, B2, or B3). The information on this genotype is considered to be exploratory (as described in Figure A4 or B4 of this document).
Topic Area: Transporters
Scenario 1: A sponsor conducts a phase 1 bioavailability study in human volunteers. The NME is a substrate of ABCB1. After the completion of the study, the sponsor genotypes the subjects for their alleles. The data may be used to explore causes of inter-individual variability in AUC.
Type of Submission:
For submissions under IND, these data could be submitted as a VGDS. For submissions under NDA/BLA, these data would be required to be submitted as a synopsis, and a VGDS of the data is encouraged.
Rationale:
The test results are not being used in decision-making or scientific arguments (such as described in Figure A1 or A2) or in the drug label or as part of the scientific database (such as described in Figure B1). In addition, polymorphism of ABCB1 is not well established. Conflicting data on the P-gp activities of various SNPs differ in published reports and is therefore neither a probable or known valid biomarker (such as described in Figures A3, B2, or B3). The information on this genotype is considered to be exploratory (as described in Figure A4 or B4 of this document).
Scenario 2: During IND development, a sponsor conducts a phase 3 clinical trial of a NME in patients with the target indication. The NME is a substrate of ABCB1. The sponsor genotypes patients for their ABCB1 alleles prior to therapy and uses two different treatment regimens based on genotypes.
Type of Submission:
Required in full report (IND)
Rationale:
The test results are used in clinical decision making (affecting dose selection) (as described in Figure A1 of this document).
Topic Area: Receptors
Scenario 1: During the IND stage of development, a sponsor reported that, based on a retrospective analysis, 5-HT1A Ser22 allele was associated with poor response to an SSRI anti-depressant. In the next clinical trial, the sponsor excludes patients with this marker genotype from the trial to enhance the drug's efficacy profile.
Type of Submission:
Required in full report (IND)
Rationale:
Data will be used in clinical decision making (entry criteria) (as described in Figure A1).
Topic Area: Clinical Outcomes- Efficacy
Scenario 1: During the IND stage of development, a sponsor of a monoclonal antibody for treatment of an autoimmune disease has discovered MHC genetic markers predictive of hypersensitivity reactions upon intravenous infusion of the product. The sponsor has also determined that serum concentrations of the antibody 4 weeks after infusion are significantly lower among patients who developed initial infusion reactions. The sponsor genotypes the MHC markers predictive of infusion reactions in every patient of a prospective clinical study. It is determined that patients with the genotypes predictive of infusion hypersensitivity (regardless of whether an infusion reaction developed or not) evidence a statistically significantly reduced response to the antibody. The sponsor proposed to highlight the improved efficacy demonstration with genetic stratification in the description of the effects of the drug.
The sponsor excludes patients with this marker genotype from the trial to enhance the drug's efficacy profile.
Type of Submission:
Required in full report (IND)
The sponsor is encouraged to develop a pharmacogenomic diagnostic test (unless it is already available), if it to be reflected in label.
Rationale:
Data will be used in clinical decision making (entry criteria) (as described in Figure A2).
Topic Area: Clinical Outcomes- Safety and Efficacy
Scenario 1: In a clinical trial, psoriatic lesions are biopsied for gene expression profiling of 160 known disease-associated genes and 140 genes potentially predictive of response for the purpose of comparing gene profiles in responders and nonresponders treated with an investigational new drug. Traditional, core clinical measurements are also made to provide evidence of efficacy and safety. The investigation is intended to identify specific gene expression patterns that could possibly be used to correlate with, and predict, efficacy or an adverse event, but at present the sponsor does not intend to incorporate the genetic information into labeling
Type of Submission:
For submissions under IND, these data could be submitted as a VGDS. For submissions under NDA/BLA, these data would be required as a synopsis, and a VGDS of the data is encouraged.
Rationale:
The test results are not being used in decision-making or scientific arguments (such as described in Figure A1 or A2). In addition, these are research data and are therefore neither a probable or known valid biomarker (such as described in Figure A3, B2, or B3). The data are considered to be exploratory (as described in Figure A4 of this document).
Scenario 2: A sponsor filed an IND 3 years ago. During clinical trials, there was lack of efficacy and so the development of the drug was abandoned. Nevertheless, the drug had some interesting pharmacological actions that warranted further investigation by the sponsor. The sponsor runs a series of genomic studies in rats and dogs with the drug and discovers a novel pharmacological profile that leads to plans to develop the drug for a different indication.
Type of Submission:
These data could be submitted as a VGDS.
Rationale:
The test results are not being used in decision-making or scientific arguments (such as described in Figure A1 or A2). In addition, these are research data and are therefore neither a probable or known valid biomarker (such as described in Figure A3). The data are considered to be exploratory (as described in Figure A4 of this document).
Scenario 2.1 Based on the results of the rat and dog pharmacogenomic studies, the sponsor, during the IND stage of development, elects to assess a subset of 25 genes in later clinical trials that may be relevant to the safety or efficacy of the compound
Type of Submission:
Required in full report (IND)
Rationale:
The sponsor is using the test results to support scientific arguments pertaining to, for example, the pharmacologic mechanism of action, the selection of drug dosing or the safety and effectiveness of a drug (as described in Figure A2.)
Topic Area: Nonclinical Safety
Scenario 1: Vasculitis is a major drug-related nonclinical safety signal and the underlying mechanism of toxicity is unknown. It is normally confirmed by histopathology. A sponsor uses new rat gene chip micro array technology to profile 8000 known genes to investigate the mechanism of toxicity and possibly see a pattern of genetic biomarkers in treated rats that is different from controls.
Type of Submission:
For submissions under IND, these data could be submitted as a VGDS. For submissions under NDA/BLA, these data would be required as a synopsis, and a VGDS of the data is encouraged.
Rationale:
The test results are not being used in decision-making or scientific arguments (such as described in Figure A1 or A2). In addition, these are research data and are therefore neither a probable or known valid biomarker (such as described in Figure A3, B2, or B3). The data are considered to be exploratory (as described in Figure A4 of this document).
Scenario 2: A sponsor filed an IND 12 months ago. During the course of subchronic toxicity testing to support longer clinical trial designs, the sponsor finds that rats develop cataracts. This finding represents a safety concern, and the sponsor elects to run toxicogenomic studies to define the mechanism of the toxicity. The sponsor discovers that the mechanism is not relevant to humans and uses the data to make their argument about human safety and the absence of cataract risk.
Type of Submission:
Required in full report (IND).
Rationale:
The sponsor is using the test results to support scientific arguments pertaining to, for example, the pharmacologic mechanism of action, the selection of drug dosing or the safety and effectiveness of a drug (as described in Figure A2)
Scenario 3: During the IND stage of development, a sponsor is investigating a new drug class and seeks to select for clinical development the best of 20 drugs showing some promise in their efficacy screen. No IND has yet been filed. The sponsor elects to assess differences in gene expression profiles to help with prioritization. The data may be generated from animal studies or from cell culture studies. The sponsor feels that the comparative profiles of gene expression alterations between the 20 drugs may help to select the most effective agent with least potential for toxicity. The data are generated to assist with compound selection and are not intended to support the safety of a proposed clinical investigation.
Type of Submission:
These data could be submitted as a VGDS (IND).
Rationale:
The test results are not being used in decision-making or scientific arguments (such as described in Figure A1 or A2). In addition, these are research data and are therefore neither a probable or known valid biomarker (such as described in Figure A3). The data are considered to be exploratory (as described in Figure A4 of this document).
Scenario 4: During the IND stage of development, a sponsor completes a 2-year carcinogenicity assay in rats and finds that there is an ambiguous tumor signal generated in the kidney, a site that is generally resistant to tumor induction. The sponsor elects to prove that the event was a spontaneous event that was not drug related by dosing the same strain of rats with drug. The sponsor succeeds in showing that there is no effect of the drug on gene expression in the kidney. A positive control shows a gene expression profile that is very consistent with known pathways of carcinogenesis. The data are used to argue to regulatory authorities that the drug is safe and does not present a tumorigenic risk to humans.
Type of Submission:
Required as full report (IND).
Rationale:
The sponsor is using the test results to support scientific arguments pertaining to, for example, the pharmacologic mechanism of action, the selection of drug dosing, or the safety and effectiveness of a drug (as described in Figure A2).
Scenario 5: A sponsor conducts global gene expression analyses to assess the relationship between dose and target organ effect. Their drug is a novel acting antipsychotic agent. The sponsor has experience that indicates that the dose-limiting effect of their drug candidate will probably injure the kidneys - an insidious chronic progressive nephropathy. Using pharmacogenomic analyses, the sponsor finds that reliable and reproducible effects on kidney gene expression occur in both rats and dogs at a dose that is 20-fold lower than the doses in 30-day studies causing a demonstrable histopathology lesion or changes in serum markers for renal toxicity. Insufficient information is currently available to definitively link the more sensitive dose-response changes in gene expression patterns to future changes in renal function or histopathologic lesions.
Type of Submission:
For submissions under IND, these data could be submitted as a VGDS. For submissions under NDA/BLA, these data would be required as a synopsis, and a VGDS of the data is encouraged.
Rationale:
The test results are not being used in decision-making or scientific arguments (such as described in Figure A1 or A2). In addition, these are research data and are therefore neither a probable or known valid biomarker (such as described in Figure A3, B2, or B3). The data are considered to be exploratory (as described in Figure A4 or B4 of this document).
GLOSSARY
The following definitions are for use in the processes outlined in this guidance and are not intended to be broadly applicable to the entire field.
Biological marker (biomarker): A characteristic that is objectively measured and evaluated as an indicator of normal biologic processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention.
1
Pharmacogenetic test: An assay intended to study interindividual variations in DNA sequence related to drug absorption and disposition (pharmacokinetics) or drug action (pharmacodynamics), including polymorphic variation in the genes that encode the functions of transporters, metabolizing enzymes, receptors, and other proteins
Pharmacogenomic test: An assay intended to study interindividual variations in whole-genome or candidate gene, single-nucleotide polymorphism (SNP) maps, haplotype markers, or alterations in gene expression or inactivation that may be correlated with pharmacological function and therapeutic response. In some cases, the pattern or profile of change is the relevant biomarker, rather than changes in individual markers.
Valid biomarker: A biomarker that is measured in an analytical test system with well-established performance characteristics and for which there is an established scientific framework or body of evidence that elucidates the physiologic, toxicologic, pharmacologic, or clinical significance of the test results. The classification of biomarkers is context specific. Likewise, validation of a biomarker is context-specific and the criteria for validation will vary with the intended use of the biomarker. The clinical utility (e.g., predict toxicity, effectiveness or dosing) and use of epidemiology/population data (e.g., strength of genotype-phenotype associations) are examples of approaches that can be used to determine the specific context and the necessary criteria for validation.
- Known valid biomarker: A biomarker that is measured in an analytical test system with well-established performance characteristics and for which there is widespread agreement in the medical or scientific community about the physiologic, toxicologic, pharmacologic, or clinical significance of the results
- Probable valid biomarker: A biomarker that is measured in an analytical test system with well-established performance characteristics and for which there is a scientific framework or body of evidence that appears to elucidate the physiologic, toxicologic, pharmacologic, or clinical significance of the test results. A probable valid biomarker may , not have reached the status of a known valid marker because, for example, of any one of the following reasons:
- The data elucidating its significance may have been generated within a single company and may not be available for public scientific scrutiny.
- The data elucidating its significance, although highly suggestive, may not be conclusive.
- Independent verification of the results may not have occurred.
Voluntary genomic data submission (VGDS): The designation for pharmacogenomic data submitted voluntarily to the FDA.
Figure A: Submission of Pharmacogenomic (PG) Data to an IND
Reports of pharmacogenomic investigations should be submitted to the IND in accordance with the decision tree below and in the formats indicated here or in the body of the guidance:
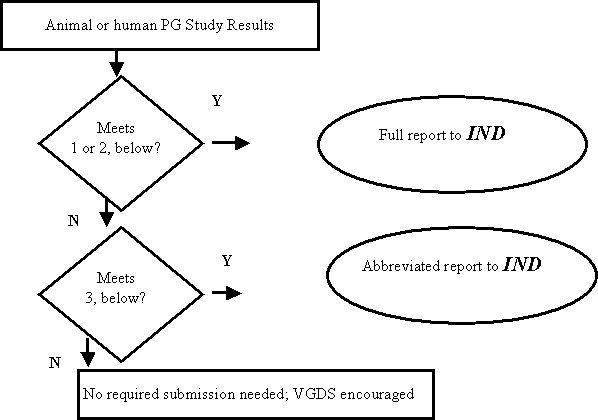
Pharmacogenomic data must be submitted to the IND under § 312.23 if ANY of the following apply:
- The test results are used for making decisions pertaining to a specific clinical trial, or in a animal trial used to support safety (e.g., the results will affect dose selection, entry criteria into a clinical trial safety monitoring, or subject stratification).
- A sponsor is using the test results to support scientific arguments pertaining to the pharmacologic mechanism of action, the selection of drug dosing or the safety and effectiveness of a drug.
- The test results constitute a known, valid biomarker for physiologic, pathophysiologic, pharmacologic, toxicologic, or clinical states or outcomes in humans, or is a known valid biomarker for a safety outcome in animal studies, or a probable valid biomarker in human safety studies. If the information on the biomarker (example, human CYP2D6 status) is not being used for purposes 1 or 2 above, the information can be submitted to the IND as an abbreviated report.
Submission to an IND is NOT needed, but voluntary submission is encouraged (i.e., information does not meet the criteria of § 312.23) if
- Information is from exploratory studies or is research data, such as from general gene expression analyses in cells/animals/humans, or single-nucleotide polymorphism (SNP) analysis of trial participants.
- Information consists of results from test systems where the validity of the biomarker is not established.
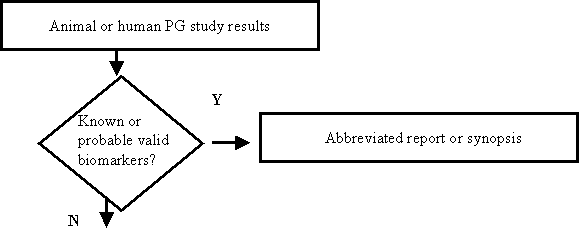
Figure B: Submission of Pharmacogenomic (PG) Data To A New NDA, BLA, or Supplement
Reports of pharmacogenomic investigations should be submitted to the NDA in accordance with the decision tree below and in the formats indicated here or in the body of the guidance:
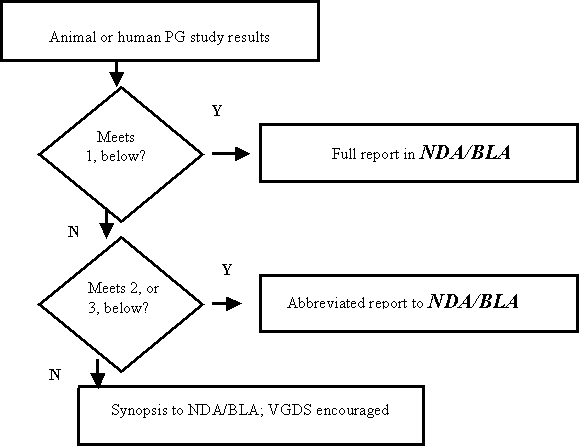
- The sponsor will use the test results in the drug labeling or as part of the scientific database being used to support approval as complete submissions (not in the form of an abbreviated report, synopsis, or VGDS), including information about test procedures and complete data, in the relevant sections of the NDA or BLA. If the pharmacogenomic test is already approved by FDA or is the subject of an application filed with the Agency, information on the test itself can be provided by cross reference.
The following examples would fit this category.
- Pharmacogenomic test results that are being used to support scientific arguments made by the sponsor about drug dosing, safety, patient selection, or effectiveness
- Pharmacogenomic test results that the sponsor proposes to describe in the drug label
- Pharmacogenomic tests that are essential to achieving the dosing, safety, or effectiveness described in the drug label
- The test results are known valid biomarkers for physiologic, pathophysiologic, pharmacologic, toxicologic, or clinical states or outcomes in the relevant species, but the sponsor is not relying on or mentioning this in the label. Submit to the Agency as an abbreviated report (not as a synopsis or VGDS). If a pharmacogenomic test of this type was conducted as part of a larger overall study, the reporting of the pharmacogenomic test results can be incorporated into the larger study report.
- The test results represent probable valid biomarkers for physiologic, pathophysiologic, pharmacologic, toxicologic, or clinical states or outcomes in the relevant species. Submit to the Agency as an abbreviated report. If the pharmacogenomic testing of this type was conducted as part of a larger study, the abbreviated report can be appended to the report of the overall study.
- Information from general exploratory or research studies, such as broad gene expression screening, collection of sera or tissue samples, or results of pharmacogenomic tests that are not known or probable valid biomarkers to the NDA or BLA are not required to be submitted. Because the Agency does not view these studies as germane in determining the safety or effectiveness of a drug, the submission requirements in §§ 314.50 or 601.2 will be satisfied by the submission of a synopsis of the study. However, the Agency encourages the voluntary submission of the data from the study in a VGDS submitted to the NDA or BLA.
Figure C: Submission of Pharmacogenomic (PG) Data to an Approved
NDA, BLA, or Supplement
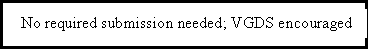
1
Biomarkers Definitions Working Group, "Biomarkers and Surrogate Endpoints: Preferred Definitions and Conceptual Framework," Clinical Pharm. & Therapeutics, vol. 69, N. 3, March 2001.
|