
(Posted: 11/29/2001)
Additions: Color green and underlined: text
addition example
Deletions: Color red and strikethrough: text
deletion example
ACCUTANE (isotretinoin) Capsules
[October 30, 2001: Hoffmann-La Roche]
[Other safety related information not appearing in the 2001 PDR: http://www.fda.gov/medwatch/safety/2001/safety01.htm#accuta]
Labeling provides for revisions to labeling to reflect the System to Manage Accutane Related Teratogenicity (S.M.A.R.T.) Program, an enhanced risk management program to help prevent fetal exposure to Accutane. In addition, this application specifies several evaluation metrics including those related to 1) female participation in the Accutane Survey conducted by the Slone Epidemiology Unit of Boston University, and 2) prescriber and pharmacist compliance with the use of Accutane qualifying stickers.
To accommodate these changes the labeling has been extensively revised. Contact the company for a copy of the new labeling/package insert
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
[October 30, 2001: Pharmacia & Upjohn]
Geriatric Use
Clinical experience with ANSAID suggests that elderly patients may have a higher incidence of gastrointestinal complaints than younger patients, including abdominal ulceration, bleeding, flatulence, bloating and abdominal pain. To minimize the potential risk for gastrointestinal events, the lowest effective dose should be used for the shortest possible duration (see WARNINGS, Gastrointestinal Effects). Likewise, elderly patients are at greater risk of developing renal decompensation (see PRECAUTIONS, Renal Effects).
The pharmacokinetics of flurbiprofen do not seem to differ in elderly patients from those in younger individuals (see CLINICAL PHARMACOLOGY, Special Populations). The rate of absorption of NSAID was reduced in elderly patients who also received antacids, although the extent of absorption was not affected (see CLINICAL PHARMACOLOGY, Drug-Drug Interactions)
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
BETAPACE (sotalol HCl) Tablets
&
BETAPACE AF (sotalol HCl) Tablets
[October 1, 2001: Berlex]
CLINICAL PHARMACOLOGY
Mechanism of Action:
In children, a Class III electrophysiologic effect can be seen at daily doses of 210 mg/m 2 body surface area (BSA). A reduction of the resting heart rate due to the beta-blocking effect of sotalol is observed at daily doses ³ 90 mg/m 2 in children.
Electrophysiology:
Twenty-five children in an unblinded, multicenter trial with supraventricular (SVT) and/or ventricular (VT) tachyarrhythmias, aged between 3 days and 12 years (mostly neonates and infants), received an ascending titration regimen with daily doses of 30, 90 and 210 mg/m 2 with dosing every 8 hours for a total of 9 doses. During steady state, the respective average increases above baseline of the QTc interval, in msec (%), were 2(+1%), 14(+4%) and 29(+7%) msec at the 3 dose levels. The respective mean maximum increases above baseline of the QTc interval, in msec (%), were 23(+6%), 36(+9%) and 55(+14%) msec at the 3 dose levels. The steady-state percent increases in the RR interval were 3, 9 and 12%. The smallest children (BSA<0.33m 2 ) showed a tendency for larger Class III effects (D QTc) and an increased frequency of prolongations of the QTc interval as compared with the larger children (BSA ³ 0.33m 2 ). The beta-blocking effects also tended to be greater in the smaller children (BSA<0.33.m 2 ). Both the Class III and beta-blocking effects of sotalol were linearly related with the plasma concentrations.
Pharmacokinetics:
The combined analysis of two unblinded, multicenter trials (a single dose and a multiple dose study) with 59 children, aged between 3 days and 12 years, showed the pharmacokinetics of sotalol to be first order. A daily dose of 30 mg/m 2 of sotalol was administered in the single dose study and daily doses of 30, 90 and 210 mg/m 2 were administered q 8h in the multi-dose study. After rapid absorption with peak levels occurring on average between 2 - 3 hours following administration, sotalol was eliminated with a mean half-life of 9.5 hours. Steady-state was reached after 1 - 2 days. The average peak to trough concentration ratio was 2. BSA was the most important covariate and more relevant than age for the pharmacokinetics of sotalol. The smallest children (BSA<0.33m 2 ) exhibited a greater drug exposure (+59%) than the larger children who showed a uniform drug concentration profile. The intersubject variation for oral clearance was 22%.
PRECAUTIONS
Pediatric Use:
The safety and effectiveness of Betapace in children have not been established.
However, the Class III electrophysiologic and beta-blocking effects, the pharmacokinetics, and the relationship between the effects (QTc interval and resting heart rate) and drug concentrations have been evaluated in children aged between 3 days and 12 year old. (See Clinical Pharmacology section.)
ADVERSE REACTIONS:
In an unblinded multicenter trial or 25 patients with SVT and/or VT receiving daily doses of 30, 90 and 210 mg/m 2 with dosing every 8 hours for a total of 9 doses, no torsade de pointes or other serious new arrhythmias were observed. One (1) patient, receiving 30 mg/m2 daily, was discontinued because of increased frequency of sinus pauses/bradycardia. Additional cardiovascular AEs were seen at the 90 and 210 mg/m 2 daily dose levels. They included QT prolongations (2 patients), sinus pauses/bradycardia (1 patient), increased severity of atrial flutter and reported chest pain (1 patient). Values for QTc ³ 525 msec were seen in 2 patients at the 210 mg/m 2 daily dose level. Serious adverse events including death, torsade de pointe, other proarrhythmias, high-degree A-V blocks and bradycardia have been reported in infants and/or children.
DOSAGE AND ADMINISTRATION
Children: As in adults the following precautionary measures should be considered when initiating sotalol treatment in children: initiation of treatment in the hospital after appropriate clinical assessment; individualized regimen as appropriate; gradual increase of doses if required; careful assessment of therapeutic response and tolerability; and frequent monitoring of the QTc interval and heart rate.
For children aged about 2 years and greater, with normal renal function, doses normalized for body surface area are appropriate for both initial and incremental dosing. Since the Class III potency in children (See Clinical Pharmacology) is not very different from that in adults, reaching plasma concentrations that occur within the adult dose range is an appropriate guide. From pediatric pharmacokinetic data the following is recommended. For initiation of treatment, 30 mg/m 2 three times a day (90 mg/m 2 total daily dose) is approximately equivalent to the initial 160 mg total daily dose for adults. Subsequent titration to a maximum of 60 mg/m2 (approximately equivalent to the 360 mg total daily dose for adults) can then occur. Titration should be guided by clinical response, heart rate and QTc, with increased dosing being preferably carried out in-hospital. At least 36 hours should be allowed between dose increments to attain steady state plasma concentrations of sotalol with age-adjusted normal renal function.
For children aged about 2 years or younger the above pediatric dosage should be reduced by a factor that depends heavily upon age, as shown in the following graph, age plotted on a logarithmic scale in months.
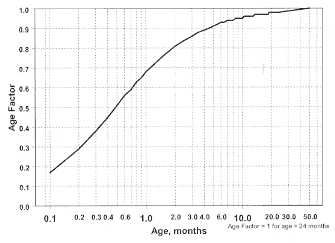
For a child aged 20 months the dosing suggested for children with normal renal function aged 2 years or greater should be multiplied by about 0.97; the initial starting dose would be (30 X 0.97)=29.1 mg/m 2 , administered three times daily. For a child aged 1 month, the starting dose should be multiplied by 0.68; the initial starting dose would be (30 X 0.68)= 20 mg/m 2 , administered three times daily. For a child aged about
1 week, the initial starting dose should be multiplied by 0.3; the starting dose would be (30 x 0.3) 9 mg/m 2 . Similar calculations should be made for increased doses as titration proceeds. Since the half-life of sotalol decreases with decreasing age (below about 2 years), time to steady state will also increase. Thus, in neonates the time to steady state may be as long as a week or longer.
In all children, individualization of dosage is required. As in adults BETAPACE (sotalol hydrochloride should be used with particular caution in children if the QTC is greater than 500 msec on therapy and serious consideration should be given to reducing the dose or discontinuing therapy when QTC exceeds 550 msec.
DOSAGE IN RENAL IMPAIRMENT [Betapace labeling only]
The use of Betapace (sotalol hydrochloride) in children with renal impairment has not been investigated. Sotalol elimination is predominantly via the kidney in the unchanged form. Use of sotalol in any age group with decreased renal function should be at lower doses or at increased interval between doses. Monitoring of heart rate and QTC is more important and it will take much longer to reach steady state with any dose and/or frequency of administration.
Preparation of Extemporaneous Oral Solution
Betapace Syrup 5 mg/mL can be compounded using Simple Syrup containing 0.1% sodium benzoate (Syrup, NF) available from Humco Laboratories as follows:
1. Measure 120 mL of Simple Syrup.
2. Transfer the syrup to a 6-ounce amber plastic (polyethylene terephthalate [PET]) prescription bottle. NOTE: An oversized bottle is used to allow for a headspace, so that there will be more effective mixing during shaking of the bottle.
3. Add five (5) Betapace 120 mg tablets to the bottle. These tablets are added intact; it is not necessary to crush the tablets. NOTE: The addition of the tablets can also be done first. The tablets can also be crushed if preferred. If the tablets are crushed, care should be taken to transfer the entire quantity of tablet powder into the bottle containing the syrup.
4. Shake the bottle to wet the entire surface of the tablets. If the tablets have been crushed, shake the bottle until the endpoint is achieved.
5. Allow the tablets to hydrate for approximately two hours.
6. After at least two hours have elapsed; shake the bottle intermittently over the course of at least another two hours until the tablets are completely disintegrated. NOTE: The tablets can be allowed to hydrate overnight to simplify the disintegration process.
The endpoint is achieved when a dispersion of fine particles in the syrup is obtained.
This compounding procedure results in a solution containing 5 mg/mL of sotalol HCl. The fine solid particles are the water-insoluble inactive ingredients of the tablets.
This extemporaneously prepared oral solution of sotalol HCl (with suspended inactive particles) must be shaken well prior to administration. This is to ensure that the amount of inactive solid particles per dose remains constant throughout the duration of use.
Stability studies indicate that the suspension is stable when stored at controlled room temperature (15º-30ºC/59º-86ºF) and ambient humidity for three (3) months.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
CEFZIL (cefprozil) Tablets & Oral Suspension
[October 1, 2001: Bristol-Myers Squibb]
CLINICAL PHARMACOLOGY:
The average AUC observed in elderly subjects (=65 years of age) is approximately 35–60% higher relative to young adults, and the average AUC in females is approximately 15–20% higher than in males.
Replaced with -
Healthy geriatric volunteers (>65 years of age) who received a single 1-g dose of
cefprozil had 35%-60% higher AUC and 40% lower renal clearance values compared with healthy adult volunteers 20-40 years of age. [Moved from PREACUTIONS: Geriatric Use subsection] The average AUC in young and elderly female subjects was approximately 15-20% higher than in young and elderly male subjects.
PRECAUTIONS
Geriatric Use
In clinical studies, when geriatric patients received the usual recommended adult doses, clinical efficacy and safety were acceptable and comparable to results in non-geriatric adult patients.
Replaced with -
Of the more than 4500 adults treated with CEFZIL in clinical studies, 14% were 65 years and older, while 5% were 75 years and older. When geriatric patients received the usual recommended adult doses, their clinical efficacy and safety were comparable to clinical efficacy and safety in non-geriatric adult patients. Other reported clinical experience has not identified differences in responses between elderly and younger patients, but greater sensitivity of some older individuals to the effects of CEFZIL cannot be excluded (see CLINICAL PHARMACOLOGY).
CEFZIL is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function.
Because elderly patients are more likely to have decreased renal function, care shouldbe taken in dose selection and it may be useful to monitor renal function. See DOSAGE AND ADMINISTRATION for dosing recommendations for patients with impaired renal function.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
[October 10, 2001: G.D. Searle]
Labeling provides for the use of Celebrex capsules for the management of acute pain in adults and the treatment of primary dysmenorrhea. Contact the company for a copy of the new labeling/package insert.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
COZAAR (losartan potassium) Tablets
&
HYZAAR (losartan potassium/hydrochlorothiazide) Tablets
[October 3, 2001: Merck]
[Other labeling changes not in 2001 PDR: http://www.fda.gov/medwatch/SAFETY/2001/aug01.htm#cozaar ]
(Cozaar)
PRECAUTIONS
Drug Interactions
As with other antihypertensive agents, the antihypertensive effect of losartan may be blunted by the non-steroidal anti-inflammatory drug indomethacin.
(Hyzaar)
PRECAUTIONS
Drug Interactions,
Losartan Potassium
As with other antihypertensive agents, the antihypertensive effect of losartan may be blunted by the non-steroidal anti-inflammatory drug indomethacin.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
CYKLOKAPRON (tranexamic acid) Injection
[October 31, 2001: Pharmacia & Upjohn ]
[Other labeling changes not in 2001 PDR: http://www.fda.gov/medwatch/SAFETY/2001/mar01.htm#cyklok ]
PRECAUTIONS
Geriatric Use
Clinical studies of CYKLOKAPRON did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function (See CLINICAL PHARMACOLOGY and DOSAGE AND ADMINISTRATION).
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
DANTRIUM (dantrolene sodium) Injection
[October 1, 2001: Procter & Gamble]
PRECAUTIONS:
Geriatric Use: Clinical studies of Dantrium Intravenous did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience had not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
Derma-Smoothe / FS (fluocinolone acetonide) Topical Oil
[October 10, 2001: Hill Dermaceuticals]
Labeling provides for the use of Derma-Smoothe/FS in pediatric patients 2 years and older for the treatment of atopic dermatitis. Contact the company for a copy of the new labeling/package insert.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
DIPROLENE AF (betamethasone dipropionate) Cream 0.05%
[October 3, 2001: Schering]PRECAUTIONS
CLINICAL PHARMACOLOGY
Sixty-seven pediatric patients ages 1 to 12 years, with atopic dermatitis, were enrolled in an open-label, hypothalamic-pituitary-adrenal (HPA) axis safety study.
DIPROLENE AF Cream was applied twice daily for 2 to 3 weeks over a mean body surface area of 58% (range 35% to 95%). In 19 of 60 (32%) evaluable patients, adrenal suppression was indicated by either a £ 5 mcg/dL pre-stimulation cortisol, or a cosyntropin post-stimulation cortisol £ 18 mcg/dL and/or an increase of < 7 mcg/dL from the baseline cortisol. Studies performed with DIPROLENE AF Cream indicate that it is in the high range of potency as compared with other topical corticosteroids.
INDICATIONS AND USAGE
DIPROLENE AF Cream is a high-potency corticosteroid indicated for relief of the inflammatory and pruritic manifestations of corticosteroid-responsive dermatoses in patients 13 years and older.
PRECAUTIONS
Information for Patients:
5. Other corticosteroid-containing products should not be used with DIPROLENE AF Cream without first talking to your physician.
Pediatric Use:
Use of DIPROLENE AF Cream in pediatric patients 12 years of age and younger is not recommended. (See CLINICAL PHARMACOLOGY and ADVERSE REACTIONS Sections.) In an open label study, 19 of 60 (32%) evaluable pediatric patients (aged 3 months – 12 years old) using DIPROLENE AF Cream for treatment of atopic dermatitis demonstrated HPA axis suppression. The proportion of patients with adrenal suppression in this study was progressively greater, the younger the age group. (See CLINICAL PHARMACOLOGY-Pharmacokinetics.)
Pediatric patients may demonstrate greater susceptibility to topical corticosteroid-induced HPA axis suppression and Cushing’s syndrome than mature patients because of a larger skin surface area to body weight ratio. The study described above supports this premise, as adrenal suppression in 9-12 year olds, 6- 8 year olds, 2-5 year olds, and 3 months – 1 year old was 17%, 32%, 38%, and 50%, respectively.
ADVERSE REACTIONS
Adverse reactions reported to be possibly or probably related to treatment with DIPROLENE AF Cream during a pediatric clinical study include signs of skin atrophy (telangiectasia, bruising, shininess). Skin atrophy occurred in 7 of 67 (10%) patients, involving all age groups from 3 months – 12 years of age.
Systemic absorption of topical corticosteroids has produced reversible hypothalamic-pituitary-adrenal (HPA) axis suppression, manifestations of Cushing's syndrome, hyperglycemia, and glucosuria in some patients.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact MedwatchDIPROSONE (betamethasone dipropionate) Ointment 0.05%
[October 3, 2001: Schering]
Labeling revisions provide results of pediatric safety studies conducted with DIPROSONE (betamethasone dipropionate) Products, in patients with atopic dermatitis, 12 years of age and younger.
CLINICAL PHARMACOLOGY
Eighty pediatric patients ages 6 months to 12 years, with atopic dermatitis, were enrolled in an open-label hypothalamic-pituitary-adrenal (HPA) axis safety study.
DIPROSONE Ointment was applied twice daily for 2 to 3 weeks over a mean body surface area of 58% (range 35% to 99%). In 15 of 53 (28%) evaluable patients, adrenal suppression was indicated by either a pre-stimulated cortisol concentration £ 5 mcg/dL pre-stimulation cortisol, or a cosyntropin post-stimulation cortisol £ 18 mcg/dL and an increase of < 7 mcg/dL from the baseline cortisol. Follow-up testing 2 weeks after study completion available for 2 of the 15 patients demonstrated a normally responsive HPA axis.
Studies performed with DIPROSONE Ointment indicate that it is in the high range of potency as compared with other topical corticosteroids.
INDICATIONS AND USAGE
DIPROSONE Ointment is a high-potency corticosteroid indicated for relief of the inflammatory and pruritic manifestations of corticosteroid-responsive-dermatoses in patients 13 years and older.
PRECAUTIONS
Information for Patients
5. Parents of pediatric patients should be
advised not to use tight-fitting diapers or plastic pants on a patient being
treated in the diaper area, as these garments may constitute occlusive dressing.
(see DOSAGE AND ADMINISTRATION).
5. Other corticosteroid-containing products should not be used with DIPROSONE Cream without first talking to your physician.
Pediatric Use DIPROSONE Ointment is not recommended in pediatric patients 12 years of age and younger. (See CLINICAL PHARMACOLOGY and ADVERSE REACTIONS Sections.)
In an open label study, 15 of 53 (28%) evaluable pediatric patients (aged 6 months – 12 years old) using DIPROSONE Ointment for treatment of atopic dermatitis demonstrated HPA axis suppression. The proportion of patients with adrenal suppression in this study was progressively greater, the younger the age group. (See CLINICAL PHARMACOLOGY - Pharmacokinetics.)
Pediatric patients may demonstrate greater susceptibility to topical corticosteroid-induced HPA axis suppression and Cushing's syndrome than mature patients because of a larger skin surface area to body weight ratio. The study described above supports this premise as adrenal suppression in 9-12 year olds, 6-8 year olds, 2-5 year olds, and 3 months – 1 year old was 17%, 27%, 29%, and 36%, respectively.
ADVERSE REACTIONS
Adverse reactions reported to be possibly or probably related to treatment with DIPROSONE Ointment during a pediatric clinical study include signs of skin atrophy (telangiectasia, thinness, shininess, bruising, loss of skin markings). Cutaneous atrophy of the face ocurred in 1/6 (17%) of infants, 2/9 (22%) of 2-5 year olds, and 2/6 (33%) of the 6-8 year olds. Non-facial atrophy occurred in 15%, 8%, and 9% of 2-5 year olds, 6-8 year olds, and 9-12 year olds, respectively.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
DIPROSONE (betamethasone dipropionate) Cream 0.05%
[October 3, 2001: Schering]
CLINICAL PHARMACOLOGY
Sixty-three pediatric patients ages 1 to 12 years, with atopic dermatitis, were enrolled in an open-label, hypothalamic-pituitary-adrenal (HPA) axis safety study.
DIPROSONE Cream was applied twice daily for 2 to 3 weeks over a mean body surface area of 40% (range 35% to 90%). In 10 of 43 (23%) evaluable patients, adrenal suppression was indicated by either a £ 5 mcg/dL pre-stimulation cortisol, or a cosyntropin post-stimulation cortisol £ 18 mcg/dL and/or an increase of < 7 mcg/dL from the baseline cortisol. Studies performed with DIPROSONE Cream indicate that it is in the medium range of potency as compared with other topical corticosteroids.
INDICATIONS AND USAGE
DIPROSONE Cream is a medium-potency corticosteroid indicated for relief of the inflammatory and pruritic manifestations of corticosteroid-responsive dermatoses in patients 13 years and older.
PRECAUTIONS
Information for Patients
5. Parents of pediatric patients should be advised not to use tight-fitting diapers or plastic pants on a patient being treated in the diaper area, as these garments may constitute occlusive dressing. (see DOSAGE AND ADMINISTRATION).
5. Other corticosteroid-containing products should not be used with DIPROSONE Cream without first talking to your physician.
Pediatric Use
Use of DIPROSONE cream, 0.05%, in pediatric patients 12 years of age and younger is not recommended. (See CLINICAL PHARMACOLOGY and ADVERSE REACTIONS Sections.)
In an open-label study, 10 of 43 (23%) evaluable pediatric patients (aged 2 years – 12 years old) using DIPROSONE Cream for treatment of atopic dermatitis for 2-3 weeks demonstrated HPA axis suppression. The proportion of patients with adrenal suppression in this study was progressively greater, the younger the age group. (See CLINICAL PHARMACOLOGY - Pharmacokinetics.)
Pediatric patients may demonstrate greater susceptibility to topical corticosteroid-induced HPA axis suppression and Cushing's syndrome than mature patients because of a larger skin surface area to body weight ratio. The study described above supports this premise, as suppression in 9-12 year olds, 6-8 year olds, and 2-5 year olds was 14%, 23%, and 30%, respectively.
ADVERSE REACTIONS
Adverse reactions reported to be possibly or probably related to treatment with DIPROSONE cream during a pediatric clinical study include signs of skin atrophy (bruising, shininess). Skin atrophy occurred in 3 of 63 (5%) patients, a 3-year old, a 5- year old, and a 7-year old.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
DIPROSONE (betamethasone dipropionate) Lotion 0.05%
[October 3, 2001: Schering]
CLINICAL PHARMACOLOGY
Twenty-five pediatric patients ages 6 to 12 years, with atopic dermatitis, were enrolled in an open-label, hypothalamic-pituitary-adrenal (HPA) axis safety study.
DIPROSONE Lotion was applied twice daily for 2 to 3 weeks over a mean body surface area of 45% (range 35% to 72%). In 11 of 15 (73%) evaluable patients, adrenal suppression was indicated by either a £ 5 mcg/dL pre-stimulation cortisol, or a cosyntropin post-stimulation cortisol £ 18 mcg/dL and an increase of < 7 mcg/dL from the baseline cortisol. Studies performed with DIPROSONE Lotion indicate that it is in the medium range of potency as compared with other topical corticosteroids.
INDICATIONS AND USAGE
DIPROSONE Lotion is a medium-potency corticosteroid indicated for relief of the inflammatory and pruritic manifestations of corticosteroid-responsive dermatoses in patients 13 years and older.
PRECAUTIONS
Information for Patients
5. Parents of pediatric patients should be advised not to use tight-fitting diapers or plastic pants on a patient being treated in the diaper area, as these garments may constitute occlusive dressing. (see DOSAGE AND ADMINISTRATION).
5. Other corticosteroid-containing products should not be used with DIPROSONE Cream without first talking to your physician.
Pediatric Use
Use of DIPROSONE Lotion, 0.05% in pediatric patients 12 years of age and younger is not recommended. (See CLINICAL PHARMACOLOGY and ADVERSE REACTIONS Sections.) In an open-label study, 11 of 15 (73%) evaluable pediatric patients (aged 6 years-12 years old) using DIPROSONE Lotion for treatment of atopic dermatitis for 2-3 weeks demonstrated adrenal suppression. (See CLINICAL PHARMACOLOGY - Pharmacokinetics.)
ADVERSE REACTIONS
Adverse reactions reported to be possibly or probably related to treatment with DIPROSONE Lotion during a pediatric study include: paresthesia (burning), erythema, erythematous rash, and dry skin. These adverse reactions each occurred in a different patient; 4% of the 25 patient population, respectively. An adverse reaction reported to be possibly or probably related to treatment in 2 different patients, 8%, of the 25 patients is puritis.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
GEREF Diagnostic (sermorelin acetate) Injection
[October 2, 2001: Serono Laboratories]
PRECAUTIONS
Geriatric Use:
Clinical studies of Geref Diagnostic did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
LOTRISONE (clotrimazole/betamethasone dipropionate) Cream
[October 3, 2001: Schering]
Labeling extensively revised. Contact the company for a copy of the new labeling/package insert.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
MAXAQUIN (lomefloxacin) Tablets
[October 24, 2001: UNIMED]
CLINICAL PHARMACOLOGY, Microbiology subsection was extensively revised in order to modify the list of microorganisms that showed in vitro activity. Contact the company for a copy of the new labeling/package insert.
[Not in 2000 PDR]
WARNINGS
Convulsions have been reported in patients receiving lomefloxacin.
No evidence of an effect of lomefloxacin on the electrical activity of the brain has been demonstrated.Lomefloxacin does not alter cerebral blood flow or cerebral glucose uptake in the CNS based on positron emission tomography.
[October 18, 2001: Eon]
PRECAUTIONS
Geriatric Use:
Clinical studies of meprobamate did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
MEVACOR (lovastatin) Tablets [October 22, 2001: Merck]
[Other labeling changes not in 2001 PDR: http://www.fda.gov/medwatch/SAFETY/2001/apr01.htm#mevaco ]
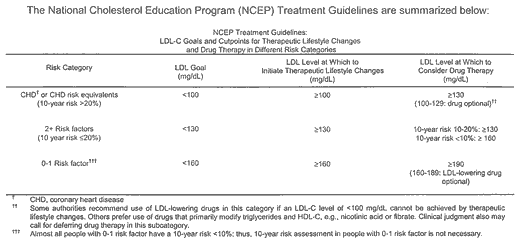
After the LDL-C goal has been achieved, if the TG is still >200 mg/dL, non-HDL-C (total-C minus HDL-C) becomes a secondary target of therapy. Non-HDL-C goals are set 30 mg/dL higher than LDL-C goals for each risk category.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
MYAMBUTOL (ethambutol) Tablets
[October 10, 2001: Lederle]
[Changes not appearing in 2000 PDR]
ADVERSE REACTIONS
The first paragraph revised and a new second paragraph added:
MYAMBUTOL may produce decreases in visual acuity which appear to be due to optic neuritis. This effect may be related to dose and duration of treatment. The effects are This effect is generally reversible when administration of the drug is discontinued promptly. In rare cases recovery may be delayed for up to one year or more. and effect may possibly be irreversible in these cases Irreversible blindness has been reported.
Optic neuropathy including optic neuritis or retrobulbar neuritis occurring in association with ethambutol therapy may be characterized by one or more of the following events: decreased visual acuity, scotoma, color blindness, and/or visual defect. These events have also been reported in the absence of a diagnosis of optic or retrobulbar neuritis.
Last sentence in the seventh paragraph revised concerning visual acuity:
Patients have then Some patients have received MYAMBUTOL ethambutol hydrochloride again after such recovery without recurrence of loss of visual acuity.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
NAVELBINE (vinorelbine tartrate) Injection
[October 2, 2001:GlaxoSmithKline]
Labeling provides for revised labeling text based on the results of a Phase 4 study conducted by the Southwest Oncology Group (SWOG 9308).
Labeling revisions, linked to the data from the trial, are extensive. These include new information from SWOG 9308 in the CLINICAL TRIALS sections, the ADVERSE REACTIONS sections, the DOSAGE AND ADMINISTRATION section. Contact the company for a copy of the new labeling/package insert.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
PARNATE (tranylcypromine sulfate) Tablets
[October 23, 2001: GlaxoSmithKline]
CONTRAINDICATIONS:
4. In combination with bupropion
The concurrent administration of a MAO inhibitor and bupropion hydrochloride (Wellbutrin, Wellbutrin SR, Zyban, GlaxoSmithKline) is contraindicated.
8. In combination with sympathomimetics
The combination of MAOIs and tryptophan has been reported to cause behavioral and neurologic syndromes including disorientation, confusion, amnesia, delirium, agitation, hypomanic signs, ataxia, myoclonus, hyperreflexia, shivering, ocular oscillations and Babinski's signs.
ADVERSE REACTIONS:
Rare instances of hepatitis, skin rash and alopecia have been reported.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
PRILOSEC (omeprazole) Capsules
[October 30, 2001: AstraZeneca]
CLINICAL PHARMACOLOGY
Pharmacokinetics and Metabolism: Omeprazole
New last paragraph -
Prilosec Delayed-Release Capsule, 40 mg was bioequivalent when administered with and without applesauce. However, Prilosec Delayed-Release Capsule, 20 mg was not bioequivalent when administered with and without applesauce. When administered with applesauce, a mean 25% reduction in Cmax was observed without a significant change in AUC for Prilosec Delayed-Release Capsule, 20 mg. The clinical relevance of this finding is unknown.
PRECAUTIONS
Information for Patients
and DOSAGE AND
ADMINISTRATION sections -
For patients who have difficulty swallowing capsules, the contents of a Prilosec
Delayed-Release Capsule can be added to applesauce. One tablespoon of applesauce should be added to an empty bowl and the capsule should be opened. All of the pellets inside the capsule should be carefully emptied on the applesauce. The pellets should be mixed with the applesauce and then swallowed immediately with a glass of cool water to ensure complete swallowing of the pellets. The applesauce used should not be hot and should be soft enough to be swallowed without chewing. The pellets should not be chewed or crushed. The pellet/applesauce mixture should not be stored for future use.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
Propylthiouracil Tablets
[October 1, 2001: Lederle]
WARNINGS
First paragraph, fourth sentence revised:
The drug should be discontinued in the presence of agranulocytosis, aplastic anemia (pancytopenia), ANCA-positive vasculitis, hepatitis, interstitial pneumonitis, fever or exfoliative dermatitis.
PRECAUTIONS:
Information for Patients
Patients should be advised that if they become pregnant during therapy or intend to become pregnant, they should contact their physician immediately about the desirability of discontinuing the drug. They also should not use propylthiouracil while nursing.
Patients should report immediately any evidence of illness, particularly sore throat, skin eruptions, fever, headache, or general malaise. They should report symptoms suggestive of hepatic dysfunction (anorexia, pruritis, right upper quadrant pain, etc).
Drug Interactions
Anticoagulants (oral): The activity of anticoagulants may be potentiated by anti-vitamin-K activity attributed to propylthiouracil.
ß-Adrenergic blocking agents: Hyperthyroidism may cause an increased clearance of beta blockers with a high extraction ratio. A dose reduction of beta-adrenergic blockers may be needed when a hyperthyroid patient becomes euthyroid.
Digitalis Glycosides: Serum digitalis levels may be increased when hyperthyroid patients on a stable digitalis glycoside regimen become euthyroid; a reduced dosage of digitalis glycosides may be required.
Theophylline: Theophylline clearance may decrease when hyperthyroid patients on a stable theophylline regimen become euthyroid; a reduced dose of theophylline may be needed.
Pediatric Use
Safety and effectiveness in pediatric patients below the age of 6 have not been established. For pediatric patients 6 years and older, see DOSAGE AND ADMINISTRATION.
ADVERSE REACTIONS:
Under "Major adverse reactions..."
Nephritis, glomerulonephritis, interstitial pneumonitis, exfoliative dermatitis, and erythema nodosum have been reported. Reports of a vasculitic syndrome associated with the presence of anti-neutrophilic cytoplasmic antibodies (ANCA) have also been received. Manifestations of ANCA-positive vasculitis may include rapidly progressive glomerulonephritis (crescentic and pauci-immune necrotizing glomerulonephritis) sometimes leading to acute renal failure; fever; pulmonary infiltrates or alveolar hemorrhage; skin ulcers; and leucocytoclastic vasculitis."
Under "Minor adverse reactions..." the following was added -
taste perversion.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
PROTONIX I.V. (pantoprazole sodium)
[October 19, 2001: Wyeth-Ayerst]
[Other labeling changes not in 2001 PDR: http://www.fda.gov/medwatch/SAFETY/2001/may01.htm#proton , http://www.fda.gov/medwatch/SAFETY/2001/jun01.htm#proton]
[Other safety related labeling changes: http://www.fda.gov/medwatch/SAFETY/2001/jul01.htm#proton2 ]
Labeling provides for the use of Protonix I.V. for Injection in the treatment of pathological hypersecretion associated with Zollinger-Ellison Syndrome (ZES).
Contact the company for a copy of the new labeling/package insert.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
Intron A (Interferon alfa-2b, recombinant) Injection
&
Rebetol (ribavirin) Capsules
[October 24, 2001: Schering]
[Other labeling changes not in 2001 PDR: http://www.fda.gov/medwatch/SAFETY/2001/mar01.htm#rebetr, http://www.fda.gov/medwatch/SAFETY/2001/feb01.htm#rebetr ]
BOXED WARNING:
Alpha interferons, including INTRON A, cause or aggravate fatal or life-threatening neuropsychiatric, autoimmune, ischemic, and infectious disorders. Patients should be monitored closely with periodic clinical and laboratory evaluations. Patients with persistently severe or worsening signs or symptoms of these conditions should be withdrawn from therapy. In many but not all cases these disorders resolve after stopping INTRON A therapy. See WARNINGS, and ADVERSE REACTIONS.
PRECAUTIONS:
Triglycerides: Elevated triglyceride levels have been observed in patients treated with interferon including REBETOL/INTRON A therapy. Elevated triglyceride levels should be managed as clinically appropriate. Severe hypertriglyceridemia (triglycerides >1000 mg/dL) may result in pancreatitis. Discontinuation of REBETOL/INTRON A therapy should be considered for patients with persistently elevated triglycerides (triglycerides >1000 mg/dL) associated with symptoms of potential pancreatitis, such as abdominal pain, nausea, or vomiting (see WARNINGS - Other).
ADVERSE REACTIONS:
The following spontaneous adverse event has also been reported during the marketing surveillance of REBETOL/INTRON A therapy: hypertriglyceridemia.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
RELAFEN (nabumetone) Tablets
[October 11, 2001: SmithKline Beecham]
PRECAUTIONS
General
Renal Effects:
Third paragraph, second, third and fourth sentences.
However, as with all NSAIDs, patients with impaired renal function should be monitored more closely than patients with normal renal function (see CLINICAL PHARMACOLOGY, Renal Insufficiency). In patients with severe renal impairment (creatinine clearance £ 30 mL/min.), laboratory tests should be performed at baseline and within weeks of starting therapy. Further tests should be carried out as necessary; if the impairment worsens, discontinuation of therapy may be warranted.
ADVERSE REACTIONS
Incidence <1%- Probably Causally Related†
Gastrointestinal: hepatic failure
Dermatologic: erythema multiforme, Stevens-Johnson Syndrome [Moved from "Incidence <1%- Causal relationship Unknown"]
Respiratory: idiopathic interstitial pneumonitis
Genitourinary: renal failure
Hematologic/Lymphatic: Thrombocytopenia [Moved from "Incidence <1%- Causal relationship Unknown"]
† Adverse reactions reported only in worldwide postmarketing experience or in the literature, not seen in clinical trials, are considered rarer and are italicized.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
RETROVIR (zidovudine)
[October 5, 2001: GlaxoSmithKline]
[Other labeling changes not in 2001 PDR: http://www.fda.gov/medwatch/SAFETY/2001/mar01.htm#retrov ]
PRECAUTIONS
Geriatric Use: Clinical studies of RETROVIR did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
RHINOCORT AQUA (budesonide) Nasal Spray
[October 26, 2001: AstraZeneca]
CLINICAL PHARMACOLOGY:
Clinical Trials:
In some studies, improvement versus placebo has been shown to occur within 24 hours of initiating treatment with RHINOCORT AQUA Nasal Spray. Maximum benefit is generally not achieved until 2 weeks after initiation of treatment.
An improvement in nasal symptoms may be noted in patients within 10 hours of first using RHINOCORT AQUA Nasal Spray. This time to onset is supported by an environmental exposure unit study in seasonal allergic rhinitis patients which demonstrated that RHINOCORT AQUA Nasal Spray led to a statistically significant improvement in nasal symptoms compared to placebo by 10 hours. Further support comes from a clinical study of patients with perennial allergic rhinitis which demonstrated a statistically significant improvement in nasal symptoms for both RHINOCORT AQUA Nasal Spray and for the active comparator (mometasone furorate) compared to placebo by 8 hours. Onset was also assessed in this study with peak nasal inspiratory flow rate and this endpoint failed to show efficacy for either active treatment. Although statistically significant improvements in nasal symptoms compared to placebo were noted within 8-10 hours in these studies, about one half to two thirds of the ultimate clinical improvement with RHINOCORT AQUA Nasal Spray occurs over the first 1-2 days, and maximum benefit may not be reached until approximately 2 weeks after initiation of treatment.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
ROWASA (mesalamine) Rectal Suspension & Suppositories
[October 1, 2001: Solvay]
ADVERSE REACTIONS:
In addition, the following adverse events have been identified during post-approval use of products which contain (or are metabolized to) mesalamine in clinical practice: nephrotoxicity, pancreatitis, fibrosing alveolitis and elevated liver enzymes. Cases of pancreatitis and fibrosing alveolitis have been reported as manifestations of inflammatory bowel disease as well. Published case reports and/or spontaneous post marketing surveillance have described rare instances of aplastic anemia, agranulocytosis, thrombocytopenia, or eosinophilia. Anemia, leukocytosis, and thrombocytosis can be part of the clinical presentation of inflammatory bowel disease.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
[October 16, 2001: DuPont]
PRECAUTIONS:
Nursing Mothers
The Centers for Disease Control and Prevention
recommend that HIV-infected mothers not breast-feed their infants to avoid risking
postnatal transmission of HIV infection. Studies in rats have demonstrated that
efavirenz is secreted in milk. Mothers should be instructed not to breast-feed
if they are receiving SUSTIVA."
The Centers for Disease Control and Prevention recommend that HIV-infected mothers not breast-feed their infants to avoid risking postnatal transmission of HIV. Although it is not known if efavirenz is secreted in human milk, efavirenz is secreted into the milk of lactating rats. Because of the potential for HIV transmission and the potential for serious adverse effects in nursing infants, mothers should be instructed not to breast-feed if they are receiving SUSTIVA.
WARNINGS
ALERT: Find out about medicines that should NOT be taken with SUSTIVA. This statement is also included on the product's bottle labels.
Concomitant use of SUSTIVA and St. John’s Wort (hypericum perforatum) or St. John’s Wort containing products is not recommended. Coadministration of non-nucleoside reverse transcriptase inhibitors (NNRTI), including SUSTIVA, with St. John’s Wort is expected to substantially decrease NNRTI concentrations and may result in sub-optimal levels and lead to loss of virologic response and possible resistance to SUSTIVA or to the class of NNRTIs.
PRECAUTIONS:
Information for Patients
A statement to patients and healthcare providers is included on the product's bottle labels: ALERT: Find out about medicines that should NOT be taken with SUSTIVA. A Patient Package Insert (PPI) for SUSTIVA is available for patient information.
SUSTIVA may interact with some drugs, therefore, patients should be advised to report to their doctor the use of any other prescription, non-prescription medication or herbal products, particularly St. John’s Wort.
Patient Package Insert (PPI):
ALERT: Find out about medicines that should NOT be taken with SUSTIVA.
Please also read the section "MEDICINES YOU SHOULD NOT TAKE WITH SUSTIVA."
Taking SUSTIVA with St. John’s Wort (hypericum perforatum) or St. John’s Wort containing products is not recommended. Talk with your doctor if you are taking or are planning to take St. John’s Wort. Taking St. John’s Wort may decrease SUSTIVA levels and lead to increased viral load and possible resistance or cross-resistance to other antiretroviral drugs.
INDICATIONS AND USAGE
Figure 3

· Proportion of patients at each time point who have HIV-RNA <500 copies confirmed by two consecutive observations and are on their original study medication and who have not experienced an AIDS-defining event.
Table 4
Study ACTG 364 - Outcomes of Randomized Treatment Through 48 Weeks*
|
Outcome |
SUSTIVA +NFV+NRTIs N=65 |
SUSTIVA +NRTIs N=65 |
NFV +NRTIs N=66 |
|
HIV-RNA < 500 copies/mL a |
71% |
63% |
41% |
|
HIV-RNA 500 copies/mL b CDC Category C event |
17% 2% |
34% 0% |
54% 0% |
|
Discontinuations for Adverse Events c Discontinuations for Other Reasons d |
3% 8% |
3% 0% |
5% 0% |
* For some patients, Week 56 data were used to confirm the status at Week 48.
a Subjects achieved virologic response (two consecutive viral loads <500 copies/mL) and maintained it through Week 48.
b Includes viral rebound and failure to achieve confirmed <500 copies/mL by Week 48.
c See ADVERSE REACTIONS for a safety profile of these regimens.
d Includes loss to follow-up, consent withdrawn, non-compliance.
WARNINGS
Psychiatric Symptoms:
Serious psychiatric adverse experiences have
been reported in patients treated with SUSTIVA. In controlled trials of 1,008
patients treated with regimens containing SUSTIVA and 635 patients treated with
control regimens, the frequency of specific serious psychiatric events among
patients who received SUSTIVA or control regimens, respectively, were: severe
depression (0.9%, 0.5%), suicidal ideation/attempts (0.5%, 0.3%), aggressive
behavior (0.3%, 0.3%), paranoid reactions (0.2%, 0.2%) and manic reactions (0.1%,
0%). Patients with a prior history of psychiatric disorders appear to be at
greater risk for these serious psychiatric adverse experiences, with the frequency
of each of the above events approximating 1%.
Psychiatric Symptoms:
Serious psychiatric adverse experiences have been reported in patients treated with SUSTIVA. In controlled trials of 1008 patients treated with regimens containing SUSTIVA for an average of 1.6 years and 635 patients treated with control regimens for an average of 1.3 years, the frequency of specific serious psychiatric events among patients who received SUSTIVA or control regimens, respectively, were: severe depression (1.6%, 0.6%), suicidal ideation (0.6%, 0.3%), non-fatal suicide attempts (0.4%, 0%), aggressive behavior (0.4%, 0.3%), paranoid reactions (0.4%, 0.3%) and manic reactions (0.1%, 0%). Patients with a history of psychiatric disorders appear to be at greater risk of these serious psychiatric adverse experiences, with the frequency of each of the above events ranging from 0.3% for manic reactions to 2.0% for both severe depression and suicidal ideation.
ADVERSE REACTIONS
Psychiatric Symptoms:
Serious psychiatric adverse experiences have
been reported in patients treated with SUSTIVA. In controlled trials the frequency
of specific serious psychiatric symptoms among patients who received SUSTIVA
or control regimens, respectively, were: severe depression (0.9%, 0.5%), suicidal
ideation or attempts (0.5%, 0.3%), aggressive behavior (0.3%, 0.3%), paranoid
reactions (0.2%, 0.2%) and manic reactions (0.1%, 0%) (see WARNINGS; Psychiatric
Symptoms). Additional psychiatric symptoms observed at a frequency of >2%
among patients treated with SUSTIVA or control regimens, respectively, in controlled
clinical trials were depression (10.0%, 8.2%), anxiety (8.2%, 5.5%), and nervousness
(5.9%, 1.9%).
Psychiatric Symptoms:
Serious psychiatric adverse experiences have been reported in patients treated withmSUSTIVA. In controlled trials the frequency of specific serious psychiatric symptoms among patients who received SUSTIVA or control regimens, respectively, were: severe depression (1.6%, 0.6%), suicidal ideation (0.6%, 0.3%), non-fatal suicide attempts (0.4%, 0%), aggressive behavior (0.4%, 0.3%), paranoid reactions (0.4%, 0.3%) and manic reactions (0.1%, 0%) (see WARNINGS; Psychiatric Symptoms). Additional psychiatric symptoms observed at a frequency of >2% among patients treated with SUSTIVA or control regimens, respectively, in controlled clinical trials were depression (15.8%, 13.1%), anxiety (11.1%, 7.6%), and nervousness (6.3%, 2.0%).
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
SYNALGOS DC (dihydrocodeine bitartrate) Capsules
[October 31, 2001: Wyeth Ayerst]
PRECAUTIONS
Geriatric Use
Clinical studies of Synalgos-DC did not include sufficient numbers of subjects aged 65 and over to determine whether elderly subjects respond differently from younger subjects.
In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
TAZORAC (tazarotene) Cream 0.1%
[October 11, 2001: Allergan]
Labeling provides for the use of Tazorac (tazarotene) Cream, 0.1%, for acne vulgaris.
Contact the company for a copy of the new labeling/package insert.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
TEQUIN (gatifloxacin) for Injection
[October 12 & 31, 2001: Bristol-Myers Squibb Company]
[Other labeling changes not in 2001 PDR: http://www.fda.gov/medwatch/SAFETY/2001/aug01.htm#tequin ]
CLINICAL PHARMACOLOGY
Glucose Homeostasis
Disturbances of blood glucose, including symptomatic hyper- and hypoglycemia, have been reported with TEQUIN (as with other quinolones), usually in diabetic patients. Therefore, careful monitoring of blood glucose is recommended when TEQUIN is administered to patients with diabetes. (See PRECAUTIONS: General, Information for Patients, and Drug Interactions .)
Microbiology
Susceptibility Tests
Diffusion techniques:
For testing Haemophilus influenzae and Haemophilus parainfluenzae a :
|
MIC (µg/mL)
|
Interpretation Susceptible (S) |
WARNINGS
Although not seen in clinical trials of TEQUIN Ruptures of the shoulder, hand, and Achilles tendons that required surgical repair or resulted in prolonged disability have been reported in patients receiving quinolones.
PRECAUTIONS
General
Because a hypotonic solution results, Water for Injection should not be used as a diluent when preparing a 2 mg/mL solution from the concentrated solution of gatifloxacin (10 mg/mL) (see DOSAGE AND ADMINISTRATION).
As with other quinolones, Disturbances of blood glucose, including symptomatic hyper- and hypoglycemia, have been reported with TEQUIN (as with other quinolones), usually in diabetic patients. receiving concomitant treatment with oral hypoglycemic agents (e.g., glyburide) with or without insulin. In these patients, The careful monitoring of blood glucose is recommended when TEQUIN is administered to patients with diabetes. If a hypoglycemic reaction occurs in or signs and symptoms of hyperglycemia occur in any patient being treated with TEQUIN, appropriate therapy should be initiated immediately and TEQUIN should be discontinued. (See CLINICAL PHARMACOLOGY, PRECAUTIONS: Drug Interactions, and ADVERSE REACTIONS.)
Information for Patients
Patients should be advised:
ADVERSE REACTIONS
Postmarketing Adverse Event Reports
The following events have been reported during postapproval use of TEQUIN. Because these events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Acute allergic reaction including anaphylactic reaction and angioneurotic edema, hepatitis, increased International Normalized Ratio (INR)/ prothrombin time, severe hyperglycemia (including hyperosmolar nonketotic hyperglycemia), severe hypoglycemia, tendon rupture, thrombocytopenia, and torsades de pointes.
DOSAGE AND ADMINISTRATION
Compatible intravenous solutions: Because a hypotonic solution results, Water for Injection should not be used as a diluent when preparing a 2 mg/mL solution from the concentrated solution of gatifloxacin (10 mg/mL) (see PRECAUTIONS).
Water for Injection,USP
|
Table 4: Gatifloxacin - Dosage Guidelines |
||
|
Infectiona |
Daily DoseB |
Duration |
|
Acute Bacterial Exacerbation of Chronic Bronchitis |
400 mg |
|
|
a due to the designated pathogens (see INDICATIONS AND USAGE). b for either the oral or intravenous routes of administration for TEQUIN (see CLINICAL PHARMACOLOGY). |
||
Patient Information About:
TEQUIN
What about other medications I am taking?
What are the possible side effects of TEQUIN?
If you have diabetes, you should know that
Disturbances of blood sugar, including high blood sugar (hyperglycemia) and
low blood sugar (hypoglycemia), have been reported in
patients treated concomitantly with TEQUIN (as with other
quinolone antibiotics), and oral antdiabetic
drugs with or without insulin usually
in diabetic patients. If you develop symptoms
of low blood sugar while on TEQUIN, you should take immediate
measures to increase your blood sugar, stop taking TEQUIN, and contact your
healthcare professional at once. If you develop high
blood sugar while on TEQUIN, you should stop taking TEQUIN, and contact your
healthcare professional at once. If you have diabetes or suspect that you
may have diabetes, discuss how to detect changes in your blood sugar with
your healthcare professional.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
TONOCARD (tocainide HCl) Tablets
[October 24, 2001: AstraZeneca]
PRECAUTIONS:
Nursing Mothers
It is not known whether tocainide is secreted in human milk. Because many drugs are secreted in human milk and because of the potential for serious adverse reactions in nursing infants from TONOCARD, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Tocainide is secreted in human milk. Because of the potential for serious adverse reactions in nursing infants from TONOCARD, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
TRILAFON (perphenazine) Tablets & Injection
[October 18, 2001: Schering]
WARNINGS
Older patients are at increased risk for development of tardive dyskinesia.
Given these considerations, especially in the elderly, antipsychotics should be prescribed in a manner that is most likely to minimize the occurrence of tardive dyskinesia.
PRECAUTIONS
Drug Interactions: Metabolism of a number of medications, including antipsychotics, antidepressants, $ - blockers, and antiarrhythmics, occurs through the cytochrome P450 2D6 isoenzyme (debrisoquine hydroxylase). Approximately 10% of the Caucasian population has reduced activity of this enzyme, so-called iopoorle metabolizers. Among other populations the prevalence is not known. Poor metabolizers demonstrate higher plasma concentrations of antipsychotic drugs at usual doses, which may correlate with emergence of side effects. In one study of 45 elderly patients suffering from dementia treated with perphenazine, the 5 patients who were prospectively identified as poor P450 2D6 metabolizers had reported significantly greater side effects during the first 10 days of treatment than the 40 extensive metabolizers, following which the groups tended to converge. Prospective phenotyping of elderly patients prior to antipsychotic treatment may identify those at risk for adverse events.
The concomitant administration of other drugs that inhibit the activity of P450 2D6 may acutely increase plasma concentrations of antipsychotics. Among these are tricyclic antidepressants and selective serotonin reuptake inhibitors, e.g.fluoxetine, sertraline and paroxetine. When prescribing these drugs to patients already receiving antipsychotic therapy, close monitoring is essential and dose reduction may become necessary to avoid toxicity. Lower doses than usually prescribed for either the antipsychotic or the other drug may be required.
Geriatric Use: Clinical studies of TRILAFON products did not include sufficient numbers of subjects aged 65 and over to determine whether elderly subjects respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients.
In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic function, concomitant disease or other drug therapy.
Geriatric patients are particularly sensitive to the side effects of antipsychotics, including TRILAFON. These side effects include extrapyramidal symptoms (tardive dyskinesia, antipsychotic-induced parkinsonism, akathisia), anticholinergic effects, sedation and orthostatic hypotension (See WARNINGS). Elderly patients taking psychotropic drugs may be at increased risk for falling and consequent hip fractures. Elderly patients should be started on lower doses and observed closely.
DOSAGE AND ADMINISTRATION
Elderly patients: With increasing age, plasma concentrations of perphenazine per daily ingested dose increase. Geriatric dosages of perphenazine preparations have not been established, but initiation of lower dosages is recommended. Optimal clinical effect or benefit may require lower doses for a longer duration. Dosing of perphenazine may occur before bedtime, if required.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
ZERIT (stavidine) Capsules & Pediatric Oral Solution
[October 16, 2001: Bristol-Myers Squibb]
Labeling changes described in January 5, 2001 Safety Letter:
http://www.fda.gov/medwatch/safety/2001/safety01.htm#zerit
WARNING
LACTIC ACIDOSIS AND SEVERE HEPATOMEGALY WITH STEATOSIS, INCLUDING FATAL CASES, HAVE BEEN REPORTED WITH THE USE OF NUCLEOSIDE ANALOGUES ALONE OR IN COMBINATION, INCLUDING STAVUDINE AND OTHER ANTIRETROVIRALS. FATAL LACTIC ACIDOSIS HAS BEEN REPORTED IN PREGNANT WOMEN WHO RECEIVED THE COMBINATION OF STAVUDINE AND DIDANOSINE WITH OTHER ANTIRETROVIRAL AGENTS. THE COMBINATION OF STAVUDINE AND DIDANOSINE SHOULD BE USED WITH CAUTION DURING PREGNANCY AND IS RECOMMENDED ONLY IF THE POTENTIAL BENEFIT CLEARLY OUTWEIGHS THE POTENTIAL RISK (SEE WARNINGS AND PRECAUTIONS: PREGNANCY).
FATAL AND NONFATAL PANCREATITIS HAVE OCCURRED DURING THERAPY WHEN ZERIT WAS PART OF A COMBINATION REGIMEN THAT INCLUDED DIDANOSINE, WITH OR WITHOUT HYDROXYUREA, IN BOTH TREATMENT-NAIVE AND TREATMENT-EXPERIENCED PATIENTS, REGARDLESS OF DEGREE OF IMMUNOSUPPRESSION (SEE WARNINGS).
WARNINGS
Lactic Acidosis/Severe
Hepatomegaly with Steatosis/Hepatic failure:
Fatal lactic acidosis has been reported in pregnant women who received the combination of stavudine and didanosine with other antiretroviral agents. The combination of stavudine and didanosine should be used with caution during pregnancy and is recommended only if the potential benefit clearly outweighs the potential risk (see PRECAUTIONS: Pregnancy)
PRECAUTIONS: Pregnancy
Second and third paragraphs added:
There are no adequate and well-controlled studies of stavudine in pregnant women. Stavudine should be used during pregnancy only if the potential benefit justifies the potential risk.
Fatal lactic acidosis has been reported in pregnant women who received the combination of stavudine and didanosine with other antiretroviral agents. It is unclear if pregnancy augments the risk of lactic acidosis/hepatic steatosis syndrome reported in nonpregnant individuals receiving nucleoside analogues (see WARNINGS: Lactic Acidosis/Severe Hepatomegaly with Steatosis/Hepatic Failure). The combination of stavudine and didanosine should be used with caution during pregnancy and is recommended only if the potential benefit clearly outweighs the potential risk. Health care providers caring for HIV-infected women receiving stavudine should be alert for early diagnosis of lactic acidosis/hepatic steatosis syndrome.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
ZINACEF (cefuroxime) Injection
[October 12, 2001: GlaxoSmithKline]
PRECAUTIONS:
General:
Cephalosporins may be associated with a fall in prothrombin activity. Those at risk include patients with renal or hepatic impairment, or poor nutritional state, as well as patients receiving a protracted course of antimicrobial therapy, and patients previously stabilized on anticoagulant therapy. Prothrombin time should be monitored in patients at risk and exogenous Vitamin K administered as indicated.
Carcinogenesis, Mutagenesis, Impairment of Fertility: Although lifetime studies in animals have not been performed to evaluate carcinogenic potential, no mutagenic activity was found for cefuroxime in the mouse lymphoma assay and a battery of bacterial mutation tests. Positive results were obtained in an in vitro chromosome aberration assay, however, negative results were found in an in vivo micronucleus test at doses up to 10 g/kg. Reproduction studies in mice at doses up to 3200 mg/kg per day (3.1 times the recommended maximum human dose based on mg/m 2 ) have revealed no impairment of fertility.
Reproductive studies revealed no impairment of fertility in animals.
Pregnancy: Teratogenic Effects: Pregnancy Category B. Reproduction studies have been performed in mice at doses up to 6400 mg/kg per day (6.3 times the recommended maximum human dose based on mg/m 2 ) and rabbits at doses up to 400 mg/kg per day (2.1 times the recommended maximum human dose based on mg/m 2 ) and have revealed no evidence of impaired fertility or harm to the fetus due to cefuroxime. There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Geriatric Use: Of the 1914 subjects who received cefuroxime in 24 clinical studies of ZINACEF, 901 (47%) were 65 and over while 421 (22%) were 75 and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater susceptibility of some older individuals to drug effects cannot be ruled out. This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function (see DOSAGE AND ADMINISTRATION).
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
ZOLOFT (sertraline HCl) Tablets & Oral Concentrate
[October 12, 2001: Pfizer]
[Other labeling changes not in 2001 PDR: http://www.fda.gov/medwatch/SAFETY/2001/aug01.htm#zoloft ,
http://www.fda.gov/medwatch/SAFETY/2001/feb01.htm#zoloft ]
PRECAUTIONS
Pediatric Use–The efficacy of ZOLOFT for the treatment of obsessive-compulsive
disorder was demonstrated in a 12-week, multicenter, placebo-controlled study
with 187 outpatients ages 6-17 (see Clinical Trials under CLINICAL PHARMACOLOGY).
The effectiveness efficacy
of ZOLOFT in pediatric patients with major depressive disorder,
or panic disorder or
PTSD has not been systematically evaluated.
The safety of ZOLOFT use in children and adolescents, ages 6-18, was evaluated in a 12-week, multicenter, placebo-controlled study with 187 outpatients, ages 6-17, and in a flexible dose, 52 week open extension study of 137 patients, ages 6-18, who had completed the initial 12-week, double-blind, placebo-controlled study. ZOLOFT was administered at doses of either 25 mg/day (children, ages 6-12) or 50 mg/day (adolescents, ages 13-18) and then titrated in weekly 25 mg/day or 50 mg/day increments, respectively, to a maximum dose of 200 mg/day based upon clinical response. The mean dose for completers was 157 mg/day. In the acute 12 week pediatric study and in the 52 week study, ZOLOFT had an adverse event profile generally similar to that observed in adults.
Sertraline pharmacokinetics were evaluated in 61 pediatric patients between
6 and 17 18
years of age with major depressive disorder and/or
OCD and revealed similar drug exposures to those of adults when plasma concentration
was adjusted for weight (see Pharmacokinetics under CLINICAL PHARMACOLOGY).
The risks, if any, that may be associated with
sertraline’s extended use in children and adolescents with OCD have not
been systematically assessed. The
risks, if any, that may be associated with Zoloft’s use beyond 1 year
in children and adolescents with OCD have not been systematically assessed.
The prescriber should be mindful that the evidence relied upon to conclude
that sertraline is safe for use in children and adolescents derives from relatively
short-term clinical studies that
were 12 to 52 weeks in duration and
from the extrapolation
of experience gained with adult patients. In particular, there are no studies
that directly evaluate the effects of long-term sertraline use on the growth,
development, and maturation of children and adolescents. Although there is
no affirmative finding to suggest that sertraline possesses a capacity to
adversely affect growth, development or maturation, the absence of such findings
is not compelling evidence of the absence of the potential of sertraline to
have adverse effects in chronic use.
Return to Quick Reference | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
HTML by JLW