


Guidance for Industry
Biological Product Deviation Reporting for Licensed Manufacturers of Biological Products Other than Blood and Blood Components
Additional copies of this guidance document are available from the Office of Communication, Training and Manufacturers Assistance, (HFM-40), 1401 Rockville Pike, Suite 200N, Rockville, MD 20852-1448, or by calling 1-800-835-4709 or 301-827-1800, or from the Internet at http://www.fda.gov/cber/guidelines.htm.
or
Office of Training and Communication, Division of Drug Information, HFD-240, Center for Drug Evaluation and Research, 5600 Fishers Lane, Rockville, MD 20857, or by calling 301- 827-4573, or from the Internet at http://www.fda.gov/cder/guidance/index.htm.
For questions on the content of this guidance, contact (CBER) Office of Compliance and Biologics Quality at 301-827-6220 or by email at bp_deviations@fda.hhs.gov; or (CDER) Division of Compliance Risk Management and Surveillance, at 301-827-8920.
U.S. Department of Health and Human Services
Food and Drug Administration
Center for Biologics Evaluation and Research
Center for Drug Evaluation and Research
October 2006
- INTRODUCTION
- BACKGROUND
- GUIDANCE
- Who Must Report? (21 CFR 600.14(a))
Examples of who must report: - What Do I Report? (21 CFR 600.14(b))
Biological Product Deviation Reporting Flow Chart for Licensed Manufacturers of Biological Products Other than Blood and Blood Components - When Do I Report? (21 CFR 600.14(c))
- How and Where Do I Report? (21 CFR 600.14(d) and (e))
- EXAMPLES OF REPORTABLE AND NON-REPORTABLE EVENTS BY MANUFACTURING SYSTEM
- Incoming Material
- Process Controls
- Testing
- Labeling
- Product Specifications
- Quality Control and Distribution
- REFERENCES
Contains Nonbinding Recommendations
Guidance for Industry
Biological Product Deviation Reporting for Licensed Manufacturers of Biological Products Other than Blood and Blood Components
| This guidance represents the Food and Drug Administration’s (FDA’s) current thinking on this topic. It does not create or confer any rights, for or on any person and does not operate to bind FDA or the public. You can use an alternate approach if the approach satisfies the requirements of the applicable statutes and regulations. If you want to discuss an alternative approach, contact the appropriate FDA staff. If you cannot identify the appropriate FDA staff, call the appropriate number listed on the title page of this guidance. |
- INTRODUCTION
- BACKGROUND
- Replaces the term “error and accident” with “biological product deviation”;
- More clearly describes the types of events that you must report to us;
- Limits reporting to those events that may affect the safety, purity, or potency of distributed products;
- Establishes a reporting time frame of 45 days from the date the event was discovered.
- CDER Ombudsman: 301-594-5480
- CBER Ombudsman: 301-827-0379
- GUIDANCE
- Who Must Report? (21 CFR 600.14(a))
- EVENT
You are a vaccine manufacturer who contracts with another establishment (contract filler) to perform the filling operation for your vaccine product. The filling operation was not performed in accordance with written production and process control procedures provided by you and the product fails to meet applicable specifications, which may affect the safety, purity, or potency of the product. - EVENT
You are a licensed test kit manufacturer who distributed an HBsAg test kit to a consignee. The consignee, a blood establishment, stored the product at an unacceptable temperature, which may affect the safety, purity, or potency of the product. - EVENT
You are a manufacturer who received glass vials and stoppers from a vendor. You determined that the vials and stoppers did not conform with all appropriate written procedures, which may affect the safety, purity, or potency of the final product. - EVENT
A Source Plasma establishment collected and tested a unit of Source Plasma and shipped it to you, a fractionator. The plasma establishment then discovered that the viral marker testing was incorrectly performed and subsequently retested a reserve sample of the unit. The unit retested as positive for a viral marker. - EVENT
You are a licensed manufacturer who distributed a product under appropriate shipping conditions to a distributor (not a licensed manufacturer of the product). The distributor shipped the product to a pharmacy. The product was not appropriately maintained under the labeled storage or shipping conditions, either at the pharmacy or at the distributor. - What Do I Report? (21 CFR 600.14(b))
- Either;
- Represents a deviation from current good manufacturing practice, applicable regulations, applicable standards, or established specifications that may affect the safety, purity, or potency of that product; or
- Represents an unexpected or unforeseeable event that may affect the safety, purity, or potency of that product; and
- Occurs in your facility or a facility under contract with you; and
- Involves a distributed biological product.
- When you did not distribute potentially affected products, regardless of the event.
- When you determined, prior to distributing the product, that the event did not actually affect the safety, purity, or potency of a product.
- When you detected an event and, prior to distribution, you made appropriate corrections, or reprocessed or reworked the product following appropriate procedures (which may require FDA approval).
- If your report would simply state that you were late in reporting an event to us.
- When there is a minor recordkeeping deviation that would not affect the safety, purity, or potency of the product, such as the omission of a date of review.
- If the product was not a US licensed product.
- After you distributed the product, your materials vendor informed you that materials used in the manufacturing process, such as components, drug product containers, or closures, did not meet all written specifications, and you could not have detected the deviation during your routine incoming material qualification procedures.
- After you distributed the product, your supplier of source or raw material informed you that the material did not meet all written specifications, and you could not have detected the deviation during your routine incoming material qualification procedures.
- determined that the event did not affect the safety, purity, or potency of the product,
- or
- reprocessed or reworked the product in accordance with appropriate procedures
- (which may require FDA approval), or
- otherwise corrected the problem (such as performed necessary testing if it is discovered
- that release testing was not performed), you are not required to report under 21 CFR 600.14.
- maintaining the continued safety, purity, and potency of the product,
- compliance with applicable product and establishment standards, and
- compliance with current good manufacturing practices.
- When Do I Report? (21 CFR 600.14(c))
- How and Where Do I Report? (21 CFR 600.14(d) and (e))
- EXAMPLES OF REPORTABLE AND NON-REPORTABLE EVENTS BY MANUFACTURING SYSTEM
- An incoming material specification event occurs during the receipt and acceptance of incoming materials (See Section IV. A.).
- A process control event occurs during the manufacturing process (See Section IV. B.).
- A testing event occurs during in-process or release testing (See Section IV. C.).
- A product labeling event occurs during the labeling process, which includes assuring the labeling information is correct, printing the label, and applying the label to the product (See Section IV. D.).
- A product specification event involves a product that failed to meet one or more of its final product specifications, at the time of release or at anytime during the labeled dating period (See Section IV. E.).
- A quality control and distribution event occurs during the quality control approval process or during distribution of the final product (See Section IV. F.).
- Incoming Material
- Containers or closures (e.g., stoppers) that did not conform to all appropriate written specifications or were found to be defective were used in manufacturing.
- Source or raw material that did not meet specifications or was otherwise found to be unsuitable was used in manufacturing. For example, the source or raw material:
- Was contaminated with microorganisms, such as bacteria or mold (if required to be sterile);
- Contained chemical impurities that exceed specifications;
- Contained precipitate (specification not met);
- Was subject to a testing deviation (required testing not performed or performed incorrectly);
- Tested positive for viral marker (e.g., Source Plasma, recovered plasma); or
- Was stored or shipped at incorrect temperature (e.g., lack of controlled shipment temperature for sensitive material).
- Component that did not meet specification was used for testing or processing (e.g., used out of date components).
- Material did not meet appropriate written specifications and was rejected and not used in manufacturing.
- Process Controls
- Manufacturing or processing was performed using incorrect parameters, such as
- Incorrect temperature;
- Filling was not performed according to procedures; or
- Aseptic processing was not performed according to procedures.
- Bulk or intermediate product was stored improperly, such as
- At the incorrect temperature; or
- For an excessive time.
- Manufacturing process was interrupted (e.g., due to a power outage).
- *Sanitization procedures were not performed or performed incorrectly.
- *Media fill failure or media fill performed incorrectly.
- Equipment was not performing properly in accordance with the written procedures.
- Equipment failure.
- Bulk material did not meet specifications or was otherwise unsuitable due to:
- Contamination with microorganisms, such as bacteria or mold (if required to be sterile);
- Chemical impurities exceeding specification; or
- Presence of precipitate (specification not met).
- In-process specification not met.
- Testing
- Safety, purity, potency, sterility, identity, or stability testing was
- Not performed or not performed at required interval; or
- Performed incorrectly.
- There was no documentation of testing performed.
- Testing not performed or not documented, but the lack of testing or documentation was discovered prior to distribution; product distributed only after completion of testing and documentation; or
- Testing performed incorrectly, but investigation determines the testing was invalid and testing appropriately repeated and found acceptable prior to distribution.
- Labeling
- Package insert was incorrect, not the current approved version, or not included with product.
- Information was missing or incorrect, such as product type, lot number, storage temperature, concentration or volume, administration route.
- Product was labeled with an extended expiration date, even if the product is expected to be used within the correct dating period.
- Product was missing expiration date.
- Product was labeled with a shortened expiration date, provided the date was not shortened because the product may not meet its specifications through the entire approved dating period.
- Product Specifications
- Final product specifications were not met, such as
- Potency;
- Moisture content; or
- Preservative content.
- Final product was unsuitable, such as
- Contaminated with microorganisms, such as bacteria or mold (if required to be sterile);
- Chemical impurities exceed specification;
- Contained precipitate (specification not met); or
- Hemolyzed (reagent red blood cells).
- Component packaged with final product failed to meet specification, such as
- Bacteriostatic Water for Injection; or
- Diluent.
- Stability testing failed during the labeled dating period and under the labeled temperature storage conditions.
- Stability testing performed for conditions beyond those approved, such as a higher storage temperature or extended expiration date, and product did not pass more rigorous tests.
- Quality Control and Distribution
- Quality control procedures were not followed or not performed; or
- Distribution of product which failed to meet release criteria or was found unacceptable, for example:
- Product was inappropriately distributed due to a failure in the distribution system;
- Product was distributed prior to determining whether the product was suitable for distribution;
- Events associated with the physical distribution of the product;
- Additional information reveals product to be unacceptable.
- Product was distributed prior to completion of all required testing;
- Product was distributed prior to resolution of a manufacturing problem that may affect the safety, purity, or potency of the product;
- Product was distributed prior to release by the quality control unit;
- Product was released prior to validation of manufacturing process;
- Product was outdated at time of distribution;
- Product was shipped at incorrect temperature or with lack of assurance that controlled temperatures were maintained during shipment when controlled storage is required; or
- Product under quarantine was distributed.
- Product was shipped to the incorrect facility;
- Product is properly labeled but the shipping invoice differs from the actual shipment, (e.g., 10 vials listed on the invoice, but only 9 vials contained in the shipment); or
- Customer order was filled incorrectly (wrong product, wrong amount), provided the product was labeled appropriately.
- Biological Products; Reporting of Errors and Accidents in Manufacturing; Proposed Rule (62 FR 49642, September 23, 1997).
- Biological Products: Reporting of Biological Product Deviations in Manufacturing; Final Rule (65 FR 66621, November 7, 2000).
- Guidance for Industry; Biological Product Deviation Reporting for Blood and Plasma Establishments (October 2006).
This guidance document provides you, a licensed manufacturer of biological products other than blood and blood components, with the FDA’s current thinking related to the biological product deviation (BPD) reporting requirements. This guidance document finalizes the draft guidance of the same title dated August 2001.
FDA’s guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidances describe the FDA’s current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidances means that something is suggested or recommended, but not required.
In the Federal Register of November 7, 2000 (65 FR 66621), we, FDA, issued a final rule amending the regulation requiring licensed manufacturers of biological products to report events that represent unexpected or unforeseeable events or deviations from current good manufacturing practice (CGMP), applicable regulations, applicable standards, or established specifications that may affect the safety, purity, or potency of a product (Title 21 Code of Federal Regulations (21 CFR 600.14).1
The amended regulation at 21 CFR 600.14, effective May 7, 2001:
Under 21 CFR 600.14, you are required to report certain events associated with the manufacturing, to include testing, processing, packing, labeling, or storage, or with the holding or distribution of a licensed biological product, which may affect the safety, purity, or potency of a distributed licensed product. Safety, purity, and potency are defined in 21 CFR 600.3(p), (r), and (s).
Under 21 CFR 600.14, you should submit reports as soon as possible, but you are required to submit reports at a date not to exceed 45 calendar days from the date of discovery of information reasonably suggesting a reportable event has occurred. To facilitate reporting, we have developed a standardized reporting format that you may submit electronically, or in paper form by mail.
The amended regulation does not change any of the requirements in 21 CFR Part 211 or Part 820 for conducting investigations of any unexplained discrepancy or the failure of a lot or unit to meet any of its specifications. Those regulations require you to evaluate and investigate, as appropriate, unexplained discrepancies and failures to meet specifications, and to maintain complaint records, including records of investigations and follow-up (21 CFR 211.192, 211.198, 820.90, and 820.100). We recommend that your procedures for the investigation of any unexplained discrepancy or the failure of a lot or unit to meet any of its specifications include provisions for:
- a timely investigation;
- an appropriate corrective action plan to prevent recurrence;
- procedures to gain control of unsuitable products in a timely manner;
- appropriate disposition of all affected products (in-date and expired).
On June 30, 2003, FDA transferred some of the therapeutic biological products that had been reviewed and regulated by the Center for Biologics Evaluation and Research (CBER) to the Center for Drug Evaluation and Research (CDER). CDER now has regulatory responsibility, including premarket review and continuing oversight, over the transferred products. In regulating the products assigned to them, CBER and CDER will consult with each other regularly and whenever necessary.
Questions about the assignment of specific products to CBER or CDER should be directed to the center jurisdiction officers at:
Under 21 CFR 600.14, the manufacturer who holds the biologics license and who had control over the product when the deviation from current good manufacturing practice (CGMP), applicable regulations, applicable standards, or established specifications, or an unexpected or unforeseeable event (such a deviation or unexpected, unforeseen event is referred to hereafter as an “event”) that may affect the safety, purity, or potency occurred, must submit a report. This reporting requirement applies to licensed manufacturers of biological products including vaccines, allergenic products, therapeutics, plasma derivatives, and in vitro diagnostics (IVDs) (e.g., test kits or reagents used for donor screening).
We define “control” in 21 CFR 600.3(ii ) as having responsibility for maintaining the continued safety, purity, and potency of the product and for compliance with applicable product and establishment standards and for compliance with current good manufacturing practices (CGMP).
If you are a plasma fractionator or IVD manufacturer that collects Source Plasma or other blood components to be used as your source material for further manufacture into a finished product, you are subject to reporting under either 21 CFR 600.14 or 21 CFR 606.171, based on when the event occurs. Under 21 CFR 606.171, you are required to report events that occur during the manufacture of such source material. Under 21 CFR 600.14, you are required to report events that occur after the manufacture of such source material and during the manufacture of a finished biological product that is licensed (e.g., Immune Globulin Intravenous (Human), Reagent Red Blood Cells).
Under 21 CFR 600.14, if you manufacture unlicensed source material (except for blood or blood components) and do not use that material in a finished product, you are not required to report events that occur during manufacturing of the unlicensed source material. You may, of course, report any events to the consignees of the material.
If you manufacture a finished licensed product in which its unlicensed source material was subject to an event in manufacturing, under 21 CFR 600.14, you must report the event if the safety, purity, or potency of the final distributed product may be affected. The use of unsuitable source material would represent an unexpected or unforeseeable event as referenced in 21 CFR 600.14(b)(1)(ii).
You are not required to report events under 21 CFR 600.14, if you manufacture an unlicensed product that is subject to an investigational new drug ( IND) application. If you manufacture a licensed product that is also being studied under an IND, you are required to report events under 21 CFR 600.14.
Cooperative Manufacturing Arrangements
Occasionally, a manufacturer establishes a contract with another entity to perform some or all of the manufacture of a product. Some common manufacturing steps performed under contract include testing, filling, storage, and distribution. If you contract out any manufacturing step, for the purposes of 21 CFR 600.14 and as described in this guidance document, that step is performed under your control. Under 21 CFR 600.14(a), you must establish, maintain and follow a procedure for receiving information from that contract manufacturing facility on all deviations, complaints, and adverse events that may affect your product.
If you are a contract manufacturer (i.e., if, under contract, you perform a step in manufacturing for another facility), you must conduct such manufacturing in accordance with CGMP (21 USC 351(a)(2)(B)), but you are not required to report BPDs to us.
Divided manufacturing is an arrangement in which two or more manufacturers, each registered with us in accordance with Part 207 or 607 and licensed to manufacture a specific biological product in its entirety, participate jointly in the manufacture of the product.
Shared manufacturing is an arrangement in which two or more manufacturers are licensed and responsible for different, specific aspects of the manufacture of a product, but neither is licensed for all aspects of the product manufacturing. A participating manufacturer may perform a particular manufacturing step and/or contract with another entity or entities to do so and assume responsibility for compliance with the applicable product and establishment standards as described for an applicant in 21 CFR 600.3(t). A participating manufacturer that performs (or is responsible for the performance of) significant product manufacturing is considered eligible for separate licensure under this arrangement.
If you are involved in divided or shared manufacturing arrangements you are responsible for reporting events that occur during the manufacturing operation(s) while the product is under your control.
Examples of who must report:
REPORTING
Under 21 CFR 211.192 and 211.198, the contract filler must perform an investigation. The contract filler would provide you with the details of the deviation that occurred during the filling process. The contract filler is NOT required to report to us.
Under 21 CFR 600.14(a), you must establish, maintain, and follow a procedure for receiving information from the contract filler about deviations concerning the filling operation. Under 21 CFR 600.14(b), you must submit a report to us if you distributed the improperly filled product. We recommend that you assure that the contract filler performed an adequate investigation.
REPORTING
You are NOT required to report to us or perform an investigation, since the product was not in your control at the time the event occurred.
Under 21 CFR 606.100(c), 211.192 and 211.198, the consignee is required to perform an investigation if they used the test kit for blood donor testing. Under 21 CFR 606.171, the consignee would submit a report to us, if they distributed blood components that were tested using the improperly stored test kit (see footnote 1).
REPORTING
We recommend that you notify the vendor so the vendor can investigate the deviation. The vendor is NOT required to report to us.
You are NOT required to report to us unless you used the unsuitable vials and stoppers and distributed the final product.
REPORTING
Under 21 CFR 600.14, you are required to submit a report if you used the improperly tested plasma in the manufacture of a licensed biological product and distributed the final product, because the safety, purity, or potency of the final product may be affected. We recommend that you establish a procedure for receiving information from the Source Plasma establishment about deviations.
Under 21 CFR 606.100(c), the plasma establishment must perform an investigation of the improper testing and release of the unit. Under 21 CFR 606.171, the plasma establishment is required to submit a report to us (see footnote 1).
REPORTING
You are not required to submit a report because the event occurred after the product left your control. The distributor would only be required to report if the distributor were licensed.
Under 21 CFR 600.14(b), you must report any event and any information relevant to the event associated with the manufacturing, to include testing, processing, packing, labeling, or storage, or with the holding or distribution, of a licensed biological product, if that event meets all the following criteria:
An adequate procedure for reporting would include steps for determining whether or not an event is one for which a report must be submitted. The decision to report should be based on whether the event had the potential to affect the safety, purity, or potency of a product. The terms safety, purity, and potency are defined in 21 CFR 600.3(p), (r), and (s), respectively.
Retrieval and Consignee Notification
You are not required to submit a BPD report simply because you failed to follow your own internal procedures for retrieval or notification (e.g., you do not have to submit a report if you did not notify consignees within the time frame prescribed in your procedures). The failure to follow retrieval or notification procedures does not, by itself, affect the safety, purity, or potency of the product. However, under 21 CFR 600.14(b), you must submit a report if the underlying reason for the retrieval or notification meets the reporting criteria found in Section III.B. In that case, the report must describe the event that may have affected the safety, purity, or potency of the distributed product and should describe the failure to follow procedures. For example, you distributed a product that did not meet specifications, which may affect the safety, purity, or potency of the product. Your standard operating procedure (SOP) requires that you notify consignees of the product within 5 days, and you notified the consignees 2 weeks after discovering the event. You are required to report the improper release of the product. We recommend that you also include in the report that consignee notification was not in accordance with your SOP.
You are not required to submit a report:
While the above examples would not be reportable under 21 CFR 600.14, the events may constitute deviations from the regulations, which we would assess during inspection.
Biological Product Deviation Reporting Flow Chart for Licensed Manufacturers of Biological Products Other than Blood and Blood Components
You may use the following flow chart to help you determine if you are required to report an event to us.
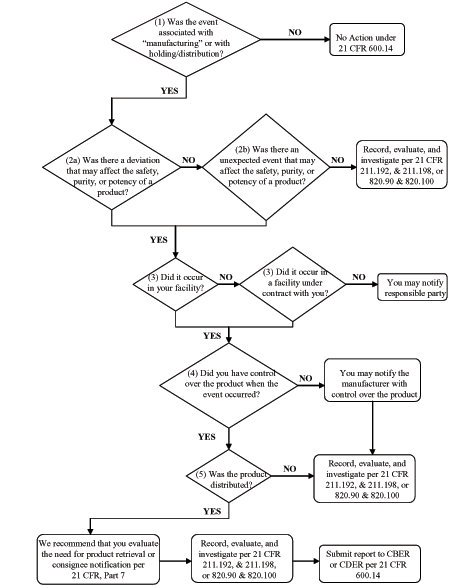
The following questions correspond to the flow chart:
(1) Was the event associated with “manufacturing” or holding or distribution as they are described in the regulation?
As described in 21 CFR 600.14(b), manufacturing includes testing, processing, packing, labeling, or storage. In addition you must report events associated with the holding or distribution, of a licensed biological product.
Under 21 CFR 600.14, you are not required to report events that occur after you distribute the product, including those that occur due to misuse or mishandling of the product by the user, such as administration errors.
Other provisions require you to report adverse experiences. Manufacturers of biological drug products are required to report adverse experiences in accordance with 21 CFR 600.80; in addition, certain vaccine adverse experiences are reportable under the Vaccine Adverse Event Reporting System (VAERS) (42 USC 300aa-25; see also guidance on www.fda.gov/cber/vaers/vaers.htm). Device manufacturers are subject to Medical Device Reporting in accordance with 21 USC 360i and 21 CFR Part 803. If a reportable adverse experience occurs in connection with a reportable deviation or unexpected or unforeseeable event in manufacturing, you must submit both an adverse experience/event report and a biological product deviation report.
(2a) Was there a deviation that may affect the safety, purity, or potency of a product?
A deviation that may affect the safety, purity, or potency of a product could include any change from the validated manufacturing process that prevents a product from meeting all CGMP requirements, applicable standards, and established specifications. The CGMP and applicable regulations are currently found in 21 CFR Parts 210, 211, 600 - 680, and 820. Standards refer to specifications and procedures applicable to the manufacture or release of products, which are established in the biologics license application and designed to help ensure the continued safety, purity, and potency of such products. Specifications refer to quality standards (i.e., tests, analytical procedures, and acceptance criteria) that confirm the quality of products and other materials used in the production of a product.
(2b) Was there an unexpected or unforeseeable event that may affect the safety, purity, or potency of a product?
An unexpected or unforeseeable event is one in which, despite the fact that you followed all required procedures, something occurred that may affect the safety, purity, or potency of a product. This may be due to information that you did not have at the time of manufacturing. Examples of unexpected or unforeseeable events in which the safety, purity, or potency of the product may be affected include the following:
If an event occurred, but could not have affected the safety, purity or potency of a product, you must record, evaluate, and investigate it in accordance with 21 CFR 211.192 and 211.198 for drug products and 21 CFR 820.90 and 820.100 for device products, but you do not need to report to us.
Most deviations and discrepancies should be identified during your manufacturing process or as part of your QC (Quality Control) activities and prior to the distribution of products. However,under 21 CFR 600.14, if you discover an event after you distributed a product and the safety, purity, or potency of the product may have been affected because of the event, you must report the event regardless of whether consignee notification or product retrieval is necessary. You must report the event even if you ultimately determined, through investigation, that the safety, purity, or potency of the product was not actually affected.
For example, if you distributed a product that was not tested for all established specifications, you must report that to FDA, even if you subsequently tested the product and found it to be acceptable.
If you discovered an event prior to distribution of a product and
For example, if you discovered a deviation in testing prior to the distribution of a product and the investigation determines the testing was invalid, and you appropriately retested the product and found it to be acceptable, you are not required to report to us under 21 CFR 600.14.
(3) Did it occur in your facility or in a facility under contract with you?
You are required to submit a report if the event occurs within your facility or a facility under contract with you, such as a testing laboratory or contract filler. Under 21 CFR 600.14(a) and (b), you must report events that occur at the contractor and you must establish, maintain, and follow a procedure for receiving information from the contract facility on all deviations, complaints, and adverse events concerning your potentially affected product.
If you are a contract manufacturer, such as a testing laboratory or contract filler, who is not a licensed manufacturer of the product, and an event occurs within your facility, you would notify the licensed manufacturer with control over the product (the entity you are under contract with), if the event may effect the safety, purity, or potency of the product. You are not responsible for reporting the event to us.
If you, a licensed manufacturer, detect an event that occurred with respect to a licensed product produced by another licensed manufacturer, you are not required to report to us. For example, if you receive licensed bulk material that was shipped under improper conditions, we recommend that you notify that manufacturer, who would be responsible for reporting to us, if appropriate. You are not required to report to us unless you used the unacceptable bulk material for manufacturing into a final product and distributed that product.
(4) Did you have control over the product when the event occurred?
You have control over the product if you have overall responsibility for:
You are responsible for reporting if you had control over the product when the event occurred and you distributed the affected product.
You have control over the product if you are the license holder and you contract with another entity to perform all or some of the manufacture of a product. Under 21 CFR 600.14(a)(1), you must establish, maintain, and follow a procedure for receiving information from the contract manufacturing facility on all deviations, complaints and adverse events. The contract manufacturer is responsible for documenting, recording, evaluating, and investigating the event under 21 CFR 211.192 and 211.198 for drug products, or 21 CFR 820.90 and 820.100 for device products. The contract manufacturer is not responsible for reporting to us.
(5) Was the product distributed?
We define “distributed” in 21 CFR 600.3(hh) as meaning that the biological product has left the control of the licensed manufacturer.
If you distributed the product, we recommend that you also assess the need for product retrieval or consignee notification in accordance with 21 CFR Part 7. You must document, evaluate, and investigate the event in accordance with 21 CFR 211.192 and 211.198 for drug products, or 21 CFR 820.90 and 820.100 for device products, regardless of whether you distributed the product.
Under 21 CFR 600.14(c), you should report a biological product deviation as soon as possible, but you must report at a date not to exceed 45 calendar days from the date that you, your agent, or another person who performs a manufacturing, holding, or distribution step under your control, acquire information reasonably suggesting that a reportable event has occurred. You acquire such information when any employee of your facility, not just those involved in quality assurance or quality control, learns about the event. As soon as you acquire information, you should make an assessment of whether the event had the potential to affect the safety, purity, and potency of products and determine the status of the products (i.e., whether they have been distributed or need to be quarantined).
If you contract with a facility to perform a manufacturing step and an event occurs at the contractor, the time period for reporting starts when your contractor learns about the event.
Under 21 CFR 600.14(d) and (e), you must use Form FDA-3486 to report BPDs. You must submit the completed report either electronically through CBER’s web site at http://www.fda.gov/cber/guidelines.htm or by mail to:
Director, Office of Compliance and Biologics Quality (HFM-600)
Center for Biologics Evaluation and Research
1401 Rockville Pike, Suite 200N
Rockville, Maryland 20852-1448
Reports involving biological therapeutic products that have been assigned to CDER should be sent to:
Division of Compliance Risk Management and Surveillance (HFD-330)
Center for Drug Evaluation and Research
Food and Drug Administration
5600 Fishers Lane
Rockville, MD 20857
If the event occurred at your contract manufacturer, we recommend that you include in the BPD Report the details reported to you by the contract manufacturer regarding the event.
Complete a separate report for each event. If an event involves more than one product, you only need to complete one report listing all distributed products affected.
The Form FDA-3486 and instructions for completing both formats may be found at http://www.fda.gov/cber/guidelines.htm.
We categorize BPD reports according to the manufacturing system where the event occurred. Following the bulleted list, each system is explained further and we provide examples of both reportable and non-reportable events. An event may be the result of a failure within a variety of systems. It is important for you to know both where the event occurred and why your product was allowed to continue through the manufacturing process and distributed, so that you can implement appropriate corrective action to prevent recurrence.
The following examples of events are not all-inclusive and do not represent all variations that may occur. The examples include deviations from CGMP, applicable regulations, applicable standards, and established specifications, in addition to unexpected or unforeseeable events. Not all of these examples will necessarily apply to you or to all products you manufacture. Whether or not the examples apply to you depends on your manufacturing operations and procedures. Under 21 CFR 211.192 and 211.198 for drug products and 21 CFR 820.90 and 820.100 for device products, all events must be investigated.
Incoming materials include, but are not limited to, source material, raw material, components, containers, and closures used in manufacturing. An event involving incoming material occurs when you receive, accept, and manufacture licensed products from incoming materials that are defective, do not meet established specifications, or otherwise are determined to be unsuitable, which you did not discover until after you distributed the product. This includes receiving information from the material supplier that the product was manufactured using non-conforming material.
Under 21 CFR 600.14(b), you must submit a report when there is an event (a deviation or unexpected or unforeseeable event) involving incoming material specifications that may affect the safety, purity, or potency of a licensed product you distributed. Examples of reportable events associated with incoming material specifications may include:
DO NOT REPORT:
Process control events occur during the manufacturing process, which you did not discover until after you distributed the product.
Under 21 CFR 600.14(b), you must submit a report when there is an event (a deviation or unexpected or unforeseeable event) during the manufacturing process that may affect the safety, purity, or potency of a licensed product you distributed. Examples of reportable events associated with the manufacturing process may include:
* If an event occurs in a process control operation that you perform only periodically, your report should identify all potentially affected products distributed since last successful operation.
Testing events include those that occur during testing, which you did not discover until after you distributed the product. This includes testing that was not performed or was performed incorrectly. It includes situations where there is no record of testing and the safety, purity, or potency of the product may be affected. This also includes testing performed using incorrect or inappropriate (e.g., expired) samples, reagents, or media.
Under 21 CFR 600.14(b), you must submit a report when there is an event (a deviation or unexpected or unforeseeable event) during testing that may affect the safety, purity, or potency of a licensed product you distributed. Examples of reportable events during testing may include:
DO NOT REPORT:
Labeling events include those that occur during the labeling process, which you did not discover until after you distributed the product. Labeling events include incorrect, missing or misleading information on any labeling pertaining to the product, including the unit label, the package insert, carton labels, and any other labeling accompanying the product.
Under 21 CFR 600.14(b), you must submit a report when there is an event (a deviation or unexpected or unforeseeable event) during labeling that may affect the safety, purity, or potency of a licensed product you distributed. Examples of reportable events associated with labeling may include:
DO NOT REPORT:
Product specification events involve a product that does not meet its specifications, either at the time of distribution or at any time during the labeled shelf life of the product, which you did not discover until after you distributed the product. Product may have been inappropriately or incorrectly analyzed during release testing, or may have deteriorated over time. These events may be discovered during an audit, during investigation into a consumer complaint, or during stability testing.
Under 21 CFR 600.14(b), you must submit a report when there is an event (a deviation or unexpected or unforeseeable event) associated with product specifications that may affect the safety, purity or potency of a licensed product you distributed. Examples of reportable events associated with product specifications may include:
DO NOT REPORT:
Events in quality control and distribution are those in which
Under 21 CFR 600.14(b), you must submit a report when there is an event (a deviation or unexpected or unforeseeable event) in the distribution process that may affect the safety, purity, or potency of a licensed product you distributed. Examples of reportable events associated with distribution, including quality control/quality assurance, may include:
DO NOT REPORT:
1See separate guidance for reporting of biological product deviations by licensed manufacturers of blood and blood components, unlicensed registered blood establishments, and transfusion services (Guidance for Industry: Biological Product Deviation Reporting for Blood and Plasma Establishments, October 2006). This guidance document is available through the Internet at http://www.fda.gov/cber/guidelines.htm or through CBER’s Office of Communication, Training, and Manufacturers Assistance.