 |
Prions: Puzzling Infectious Proteins
Ruth Levy Guyer, Ph.D.
Sometimes a scientific discovery shakes the confidence of scientists, making them question whether they truly understand nature's "ground rules."
That's exactly what prions have done to scientists' understanding of the ground rules for infectious diseases. Prions cause diseases, but they aren't viruses or bacteria or fungi or parasites. They are simply proteins, and proteins were never thought to be infectious on their own. Organisms are infectious, proteins are not. Or, at least, they never used to be.
Prions entered the public's consciousness during the mad cow epidemic that hit England in 1986. For decades, however, scientists had searched for unusual, atypical infectious agents that they suspected caused some puzzling diseases that could not be linked to any of the "regular" infectious organisms. One possibility was that slow viruses--viruses that spent decades wreaking havoc in their hosts--might be the culprits, and these putative viruses were not only leisurely about multiplying but also hard to isolate. Now researchers are coming around, albeit reluctantly, to accepting the shocking fact that naked proteins can be infectious. "More than one protein chemist has declared this to be insane--and yet this is precisely what is implied by a growing number of studies" was the way one news article put it (1).
Prions (pronounced pree-ahns) enter cells and apparently convert normal proteins found within the cells into prions just like themselves. The normal cell proteins have all the same "parts" as the prions--specifically the same amino acid building blocks--but they fold differently. They are much like the toy "Transformers" that intrigued little kids in the 1980s. A sphynx could become a robot; a bug could become a warrior. Nothing was added; nothing subtracted.
  
Prions enter brain cells and there convert the normal cell protein PrPC to the prion form of the protein, called PrPSC. When normal cell proteins transform into prions, amino acids that are folded tightly into alpha helical structures relax into looser beta sheets. More and more PrPC molecules transform into PrPSC molecules, until eventually prions completely clog the infected brain cells. The cells misfire, work poorly, or don't work at all. In mad cow disease, for example, with their brain cells running amuck, the mad cows wobble and stagger and appear fearful--their "madness" is craziness, not anger. Sheep and goats with the disease scrapie, which is like mad cow disease, become so uncomfortable and itchy that they frantically rub up against anything they can, finally scraping off--hence, the name of the disease--most of their wool and hair (2).
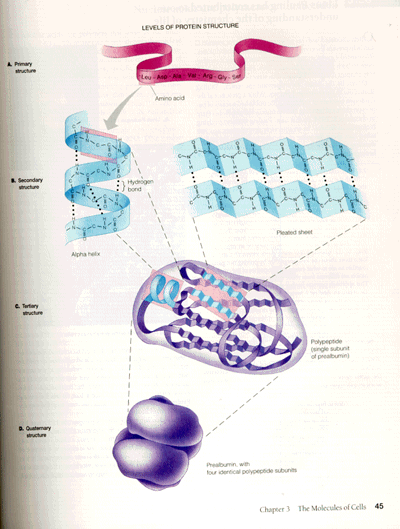
Proteins consist of stretches of amino acids (A). Some fold into alpha helices; others fold into beta pleated sheets (B). These shapes are maintained by hydrogen bonds (indicated by dots). The sheets and helices then fold together further to form the protein's tertiary structure (C). A protein, such as the one illustrated here, can include both alpha helices and beta sheets and can consist of several subunits that combine to form the quaternary structure (D). When PrPC molecules transform into PrPSC molecules, sections of the protein that were folded as alpha helices open out into beta sheets. This illustration of the levels of protein structure is taken from BIOLOGY: CONCEPTS & CONNECTIONS by Neil A. Campbell, Lawrence G. Mitchell, and Jane B. Reece. Copyright 1994 by The Benjamin/Cummings Publishing Company, Inc. Reproduced by permission.
Ultimately, infected prion-bloated brain cells die and release prions into the tissue. These prions then enter, infect, and destroy other brain cells (2). And, as clusters of cells die, the brain stops looking like a brain and starts looking more like Swiss cheese. The medical term for the prion diseases is "spongiform encephalopathies," in acknowledgement that the sick brains (encephalo is Greek for brain; pathy is Greek for disease) are riddled with holes and have taken the form of sponges.

Shepherds and farmers whose sheep had scrapie never seemed to get scrapie themselves. So, for a long time, scientists assumed that the prions of animals did not cause infections in humans. But, between 1994 and 1996, 12 people in England came down with Creutzfeld-Jakob disease (CJD), a human prion disease whose symptoms are not unlike those of the mad cows, and all the victims had eaten beef from cows suspected of having mad cow disease. In October, 1996, scientists in England reported that the prions from ten of the British patients were remarkably like those of the mad cows and not like those of people who died of "classical" CJD (3). Scientists quickly realized that the occurrence of CJD in a dozen people 19 to 39 years old was cause for alarm, because CJD had always been rare--typically one new case might be diagnosed per million people each year--and seldom occurred in people younger than 55 (1, 3). This epidemic was something new, something extraordinary. Scientists now speculate that the prions that started out in sheep suffering from scrapie made their way into cows and then moved more recently into humans. Cattle are fed meal made from sheep "offal," the bones and other waste parts of sheep carcasses. Standard procedures for grinding up carcasses were altered in the 1970s, and the new processing methods seem not to have been adequate for destroying scrapie prions. The cattle were exposed, through the offal, to sheep prions, and the prions eventually established themselves in their cow hosts. Later, they adapted further, infecting cells of people who had eaten hamburgers from prion-bearing cows.

Scrapie is an old disease, recognized in sheep and goats for more than 250 years. In 1982, it was first identified as a prion disease. The word "prion" was coined at that time by Dr. Stanley Prusiner to indicate that this disease was caused by a "proteinaceous infectious agent." The protein that causes this and all other prion diseases is called PrPSC, which stands for prion protein of scrapie (2).
At the moment, CJD and only a handful of other human diseases have clear links to prions. But it is likely that prions will turn out to be the agents of a variety of currently enigmatic diseases in which brain cells are destroyed and the nervous system deteriorates. Alzheimer's disease and Parkinson's disease are two prime candidates.
So, a couple new ground rules now seem to govern infectious diseases. The first is that naked proteins--prions--can be infectious and can cause infectious diseases. The second and potentially more troubling is that, like other infectious agents, prions can jump species' barriers and cause deadly diseases in humans. Recently, and for the first time known, two farmers with mad cows in their herds died of CJD.
Only time will tell how big a problem the prions will be both as the agents of dreadful diseases of the human nervous system and as vectors of diseases from other species. Is the juicy hamburger or the succulent lamb chop harboring microscopic "wolves in sheep's clothing?"
Human Diseases Linked to Prions
Some of the prion diseases are labelled "infectious," like the recent CJD epidemic. Others are said to be "inherited," because the PrPC gene contains a mutation that alters one or more amino acid and promotes the formation of PrPSC molecules over PrPC molecules. A third group is said to be "sporadic," because the disease seems to pop up for no apparent reason. The same disease can develop under all three circumstances.
Creutzfeldt-Jakob Disease (CJD)
Although the current epidemic of CJD appears to be linked with mad cow disease and the transmission of sheep prions to cows and then to humans, most cases of CJD in the past 20 years arose from what doctors call "therapeutic misadventures" in which a patient who was being treated for some disease or condition developed CJD as a result of the treatment (5).
In 1974, for example, a woman received a new cornea and developed CJD. The donor of the cornea had had a prion disease. In 1977, two patients with epilepsy developed CJD. Both had undergone tests during which electrodes were implanted deep in their brains. While the electrodes were recording electrical signals in the patients' brains, they were unexpectedly leaving behind deadly prions picked up from a previous patient. In the mid 1980s, a number of people receiving hormone treatments developed CJD. The hormones had been extracted from a pool of 20,000 pituitary glands, at least one of which had come from a patient who died of CJD.
The term for these misadventures is "iatrogenic" infections. The word means, literally, "physician induced" from the Greek words iatros (physician) and genic (birth, source). All were caused by doctors or equipment or treatments that were meant to heal.
Some cases of CJD appear to be genetic, and when the disease is inherited it typically develops after age 55. When the gene that encodes PrPC has a specific mutation, the formation of PrPSC rather than PrPC is favored. A genetic predisposition for CJD may account for the high incidence of CJD in central Slovakia, where it is the fifth leading cause of death. In Chile and Israel, there are also clusters of people with CJD.
Kuru
Imagine what it must be like to have a disease whose nickname is "laughing death." That's kuru, an exotic disease confined pretty much to the Fore tribe in northern regions of New Guinea. One custom of the Fore community was to eat the brains of dead relatives, a practice that promoted transmission of disease-causing prions. When cannibalism was banned, the incidence of Kuru was greatly reduced. When the kuru prion infects nerve cells, craziness and dementia, loss of coordination, and other symptoms develop.
Fatal Familial Insomnia (FFI)
This is a disease that was first associated with a prion in 1992. As its name implies, it runs in families, prevents people from sleeping, causes motor and emotional problems, and is eventually a killer. Here, too, as in inherited cases of CJD, patients have a specific mutation in the prion gene (6).
Gerstmann-Straussler-Scheinker Syndrome (GSSS)
This disease was linked to two mutations in the prion gene in 1989. PrPSC fragments accumulate in the brain in structures called plaques. In Alzheimer's disease, similar plaques develop, but they are composed of fragments of a different protein (7).
References:
- Nature 1996, 383: 666-667.
- Scientific American 1995, 272(1): 48-57.
- Nature 1996, 383: 685-690.
- Current Opinion in Neurology 1995, 8: 286-293.
- The Lancet 1992, 340: 24-27.
- Current Topics in Microbiology and Immunology 1996, 207: 19-25.
- Current Topics in Microbiology and Immunology 1996, 207: 27-34.
|
