
Comments and suggestions regarding this draft document should be submitted within [insert] days of publication in the Federal Register of the notice announcing the availability of the draft guidance. Submit written comments to the Division of Dockets Management (HFA-305), Food and Drug Administration, 5630 Fishers Lane, rm. 1061, Rockville, MD 20852. Alternatively, electronic comments may be submitted to http://www.regulations.gov. All comments should be identified with the docket number listed in the notice of availability that publishes in the Federal Register.
For questions regarding this document contact Sally Hojvat, PhD, CDRH/OIVD/DMD, by phone at (240) 276-0711, or by email at sally.hojvat@fda.hhs.gov. For statistical inquiries, please contact Marina V. Kondratovich, PhD, CDRH/Division of Biostatistics by phone at (240) 276-3126 or by email at marina.kondratovich@fda.hhs.gov. For questions relating to devices regulated by CBER, contact the Office of Communications, Training, and Manufacturers Assistance, CBER at 301-827-1800 or 800-835-4709.
  |
U.S. Department of Health and Human Services |
Draft - Not For Implementation
Additional copies are available from the Internet http://www.fda.gov/cdrh/oivd/guidance/1660.pdf. You may also send an email request to dsmica@fda.hhs.gov to receive an electronic copy of the guidance, or send a FAX request to 240-276-3151 to receive a hard copy. Please use the document number 1660 to identify the guidance you are requesting.
Or, contact:
Office of Communication, Training, and Manufacturers Assistance, HFM-40
Center for Biologics Evaluation and Research
Food and Drug Administration
1401 Rockville Pike, Suite 200N, Rockville, MD 20852-1448
Internet: http://www.fda.gov/cber/guidelines.htm
Tel: 800-835-4709 or 301-827-1800
| This draft guidance, when finalized, will represent the Food and Drug Administration's (FDA's) current thinking on this topic. It does not create or confer any rights for or on any person and does not operate to bind FDA or the public. You can use an alternative approach if the approach satisfies the requirements of the applicable statutes and regulations. If you want to discuss an alternative approach, contact the FDA staff responsible for implementing this guidance. If you cannot identify the appropriate FDA staff, call the appropriate number listed on the title page of this guidance. |
This draft guidance presents a least burdensome regulatory approach to gain FDA approval of Class III or certain licensed1 in vitro diagnostic devices in cases when a previously approved or licensed assay is migrating (i.e., transitioning) to another system for which the assay has not been previously approved or licensed.2 In this guidance the term “New System” refers to the unapproved/unlicensed system (assay, instrument, and software) to which the assay is migrating from a previously approved/licensed system. Conversely, the term “ Old System” refers to the approved/licensed system (assay, instrument and software) from which the assay is migrating to a currently unapproved/unlicensed system.
The focus of this guidance is on the study designs and performance criteria that should be fulfilled in order for a sponsor to utilize the migration study approach in support of the change. FDA will review information from the sponsor, including results of analytical and comparison studies outlined in this guidance, as well as device descriptions and risk analyses, to determine whether the use of the approved/licensed assay with the New System may compromise safety and effectiveness of the assay. The guidance document describes information that we recommend you include in a PMA (premarket approval application) supplement or BLA (Biologics License Application). For devices regulated by OIVD, sponsors may contact OIVD, and for those regulated by CBER, sponsors may contact CBER to obtain feedback concerning study plans.
FDA's guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidances describe the Agency's current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidances means that something is suggested or recommended, but not required.
This draft guidance document reflects our careful review of what we believe are the relevant issues related to migration studies and what we believe would be the least burdensome way of addressing these issues. If you have comments on whether there is a less burdensome approach, however, please submit your comments as indicated on the cover of this document.
The FDA believes that the assay migration study paradigm discussed in this draft guidance provides a least burdensome scientific and regulatory pathway for manufacturers to transfer a previously approved or licensed assay with full clinical data from an Old System to a New System (not approved or licensed). The paradigm is suitable in cases when sufficient knowledge can be derived from the documentation of design controls, risk analyses, and prior performance studies on an Old System.
If you make further modifications or iterations of the Old or New System you should compare back to the original Old System that has full clinical data when performing new migration studies. However, if the Old System with full clinical data is no longer available please contact the appropriate FDA CDRH/CBER Division for further discussion.
The migration studies approach is related to the Replacement Reagent and Instrument Family Policy that FDA uses for many Class I and Class II diagnostic devices.3 Similar to that policy migration studies rest on the premise that as platform changes are made throughout the lifetime of an approved or licensed assay, smaller and more focused analytical and clinical data sets than have traditionally been called for, along with prior knowledge of device design and performance, could allow for credentialing of the safety and effectiveness profile of the modified system.
Use of this review approach is practical, risk based, and consistent with FDA’s Critical Path Initiative, which is intended to help bring new medical products to market successfully and efficiently4. FDA believes that with proper controls and review, migration studies will meet regulatory thresholds for premarket review in a manner that will be least burdensome for both companies and FDA while protecting public health.
This draft guidance is intended to be applied, where appropriate, to licensed donor screening tests5 and approved (Class III) in vitro diagnostic assays, as well as cleared assays for which migration to a New System presents concerns . Possible scenarios for assay migration are when the assay is being transferred from a manual system to an automated or semi-automated instrument system or from a semi-automated instrument system to a fully automated instrument system, or from one automated instrument system to another (and visa versa for all scenarios). A broad variety of methodologies may use the migration studies paradigm depending on what is known about the two Systems involved. Assay transfer may be from one approved or licensed Old System to a New System that has the same technical characteristics. However, if scientific evidence suggests migration studies may not be adequate to predict actual clinical testing performance on the New System, the assay migration paradigm should not apply and FDA will recommend that traditional evaluation studies be performed.
Assay migration studies are ideally suited for test systems for which the assay output (raw signal) is a numerical result or is expressed as a signal to cutoff (S/CO). Devices for which a numerical output is not available might be more difficult to analyze and may not be suitable candidates for use of this approach.
Migration studies would not be applicable to the following devices or to system changes that are generally considered significant, such as:
The FDA strongly recommends that sponsors discuss proposed migration studies with the Agency early in the product and testing design process in order to determine if the proposed changes are consistent with the parameters that would allow for streamlined and focused testing. For CBER, this may be through an IND, or protocol review, providing preliminary protocols, data, and justifications prior to performing the migration studies. The size, nature, and scope of migration studies we recommend will depend on a detailed evaluation of the Old and New Systems, the level of regulation appropriate for the product (e.g., PMA or BLA), the performance characteristics of the assay, and the design and scope of the analytical testing and clinical trials used to support approval or licensure of the assay on the Old System.
A sponsor should take into account the following critical considerations in determining whether the migration studies approach is appropriate for a particular product, and describe these considerations in the submission:
In addition to addressing each of the critical considerations noted in Section IV, you should also include the information listed below to demonstrate the applicability of migration studies to the transfer of the assay from the Old System to the New System. The information in your submission should include, but is not limited to, the following:
In addition, the sponsor should include d ocumentation on software and instrumentation for the New System. When appropriate for the device, this documentation should meet all recommendations for the appropriate Level of Concern.7 To better control risk, the studies should be performed on the final model and software version of the New System that is intended to be marketed.
This section outlines specific studies that may be appropriate to support assay migration for in vitro diagnostic devices. Before preparing to use the migration studies approach, you should determine whether the assay is quantitative or qualitative, according to the definitions in this guidance. Specifically, for the purposes of this guidance, qualitative assays are those that determine numerical values (e.g., signal, S/CO), which are used for categorical determination of assay results (e.g., positive or negative). Quantitative assays determine numerical values which are referenced to a linear range, and standards that allow absolute determination of analyte concentrations. Section VI.A describes studies we recommend for Qualitative assays; Section VI.B addresses Quantitative Assays. Special considerations for blood screening assays are covered in Appendix I, “Migration Studies for Blood Donor Screening Assays.”
The evaluations described below are based on the idea that similar studies were conducted previously for the Old System. If the study design of the analytical studies conducted for the Old System were different from the design of the studies described in this guidance, please contact the FDA for feedback. If you believe that some of these studies do not apply to your particular device, you should present your justification for FDA review.
We recommend that you use fresh clinical specimens for all analytical studies. If this is impractical, in some cases you may substitute or supplement fresh clinical specimens with banked samples. If banked samples are not available, spiked or diluted clinical samples may be used. In some instances, use of otherwise contrived matrix-specific samples may also be appropriate; however these should mimic clinical specimens as much as is feasible. We recommend that you contact FDA if you wish to discuss appropriate sample types for these evaluations. The matrix of any of these alternative specimens should be the same as that specified by the intended use of the Old System.
You should evaluate the performance of the assay on the New System compared to the Old System at low analyte levels with dilution panels and seroconversion panels (if applicable).
We recommend that you conduct in-house within-laboratory precision studies, (to supplement the external site reproducibility studies described below in Section c). When appropriate and justified, the in-house within-laboratory precision study may not be necessary, such as (i) if you established that the New System only needs to be recalibrated at relatively long time-intervals (e.g., 6 months or more) and any other concerns can be appropriately addressed by the reproducibility study, or (ii) if the New System is recalibrated daily, so that calibration cycle variability is inseparable from day-to-day variability (which is assessed by the reproducibility studies described below) and any other concerns can be appropriately addressed by the reproducibility study
It may be sufficient to perform within-laboratory precision studies only on the New System. However, if the study design or composition of the precision panel of the Old System precision study was very different from that described in this guidance, it may be important to perform the precision study on the Old System as well. The within-laboratory precision study described below is based on modified CLSI document EP5-A2.
Sources of variability we recommend for the within-laboratory precision study are at least 12 days of testing, with 2 runs per day, and 2 replicates of each sample per run. These 12 days are not necessarily consecutive and they should span at least two calibration cycles (the calibration cycles may be non-consecutive). For each cycle, days at the beginning and end of the cycle should be included (e.g., 3 days at the beginning and 3 days at the end of each cycle, for each cycle). You should include other additional sources of variability in the design of the study, if they are important to the specific assay (e.g., operators, lots, etc.). In such cases, overall modification to the variables might be possible (e.g., spreading days of testing between different operators). If analytical and clinical performance is similar across all matrices that are included in the intended use of the Old System, then establishing performance of the New System using the most commonly employed matrix may suffice.
In concept, a cutoff for a qualitative test is established based on acceptable clinical performance (e.g., sensitivity/specificity) for the samples from the intended use population. This cutoff (threshold) is used for defining positive and negative results of the test. When the observed result exceeds the threshold, the result is considered positive (or reactive); when the observed result is below the cutoff, it is considered negative (or not reactive). A useful characteristic of the cutoff is that a sample with an actual concentration at the cutoff yields a positive result 50% of the time and a negative result 50% of the time when a large number of replicates of that sample are run under stipulated conditions (see Figure 1 below). We denote this concentration as C50.
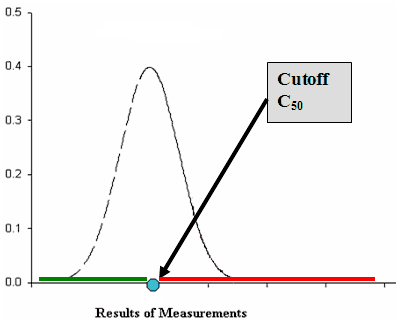
Figure 1. Results of a qualitative test for a sample with a concentration at the cutoff
For samples with concentrations exceeding C50, one expects to see positive results more than 50% of the time and similarly for samples with a concentration below C50, one expects to see positive results less than 50% of the time. In this guidance we refer to an analyte concentration that yields, upon evaluating many replicates, a positive result 95% of the time (and negative result 5% of the time) as a Low Positive concentration (C95 concentration). We refer to a sample concentration below the C50 which yields a positive result 5% of the time (and negative result 95% of the time) as a High Negative concentration (C 5 concentration). When the limit of blank (LoB) is used as a cutoff, then the concentration C95 is the same as the limit of detection (LoD) and zero concentration (no analyte present in sample) is C5. The LoB and LoD are discussed more thoroughly in Clinical Laboratory and Standards Institute (CLSI) document EP17-A.
Samples with concentrations of analyte close to C95 and C5 as determined by the Old System are recommended for the within-laboratory precision (see CLSI EP12-A2). The panel should consist of at least three members, as described below (also, see Figure 2):
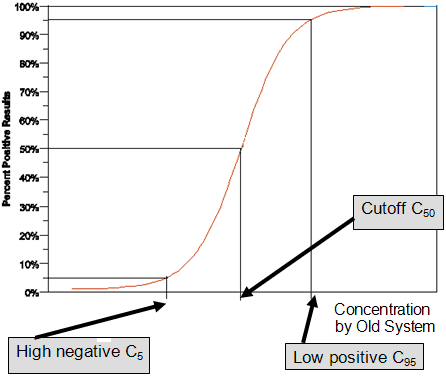
Figure 2. Relationship between percent of positive results and the analyte cutoff concentration.
For details of how the C95 and C5 concentrations can be evaluated from the previous precision studies of the Old System (see Appendix II: Statistical Notes, 1). For the precision study of the New System, it is not necessary to have the high negative and low positive samples at exactly C5 or C95 of the Old System. If the high negative and low positive samples in the precision study of the New System are close enough to the cutoff that the standard deviation (or percent coefficient of variation (CV)) is approximately constant over the range around the cutoff, the C5 and C95 of the New System can be evaluated from this precision study (see Appendix II: Statistical Notes, 1).
In addition to the high negative, low positive, and moderate positive samples, you should run the appropriate control material (negative and positive controls) and calibrators in the precision study.
We recommend you perform the reproducibility study based on a modification of CLSI EP15-A2 on the New System . The panel composition and analyte levels for this study should be the same as described in the within-laboratory precision study (Section A.1.b) and sources of variability should include testing for at least 5 days, 2 runs per day, with 3 replicates of each panel member per run at 3 laboratories (1 in-house and 2 external sites). Other sources of variability might be applicable if relevant to the specific assay (e.g., operators). If analytical and clinical performance is similar across all matrices that are indicated in the intended use of the Old System, then establishing performance of the New System using the most commonly employed matrix may suffice.
For each concentration level, similar information should be available for the Old System. Otherwise, you should perform a new reproducibility study for the Old System with study design and concentration levels as described in this section.
You should perform comparison studies using comparison panels. However, minor changes to the Old System might not warrant performing all comparison studies. The extent of the utility of these studies can be evaluated on a case-by-case basis in consultation with the FDA.
For each analyte, the qualitative assay comparison panels should consist of the following:
You should test the comparison panels on the Old System at a minimum of one site. This may be done in-house. However, you may want to use more than one Old System to better assess instrument bias. The New System should be tested at a minimum of three sites (one may be in-house) with at least one reagent kit lot. Each panel member should be tested at least four times: once with the Old System and three times with the New System. You should send the same positive and negative panels to each site, rather than dividing the panel between the three sites. Three different builds of the New System should be tested, one at each of the three sites.
We recommend you include the information described in this section for each of the respective studies, in your submission:
In addition to the acceptance criteria for performance of the New System at low levels of analyte and for seroconversion panels (if applicable), we recommend you apply the following criteria to demonstrate that there are no changes to performance characteristics that could affect the safety and effectiveness of the device:
If you believe that some of these studies do not apply to your particular device, you should describe your reasoning in detail in your application to FDA. If the design of the analytical studies conducted for the Old System were different from those described in this guidance, please contact FDA.
We recommend that you use fresh clinical specimens for all analytical studies. If this is impractical in some cases you may substitute or supplement fresh clinical specimens with banked samples. If banked samples are not available, spiked or diluted clinical samples may be used. In some instances, use of otherwise contrived matrix-specific samples may also be appropriate, however these should mimic clinical specimens as much as is feasible. We recommend that you contact FDA if you wish to discuss appropriate sample types for these evaluations. The matrix of any of these alternative specimens should be the same as that specified by the intended use of the Old System.
For assays that were previously approved or licensed with a specified LoB (limit of blank) and LoD (limit of detection), the same evaluations should be repeated with the New System. The study should demonstrate that the LoB and LoD are very similar for both systems (a protocol is described in CLSI EP17-A). Specifically, the sample with a concentration at the LoD (reported as “analyte detected” approximately 95% of the time, measured by the Old System) should also be reported as “analyte detected” approximately 95% of the time, if measured by the New System (see Figure 3 below).
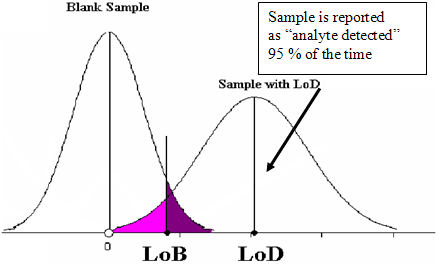
Figure 3. Relationship Between Measurements of the Blank Sample and Limit of Detection Sample.
The limit of quantification (LoQ, or lower limit of measuring range) of the New System should be estimated and compared with the LoQ of the Old System (see CLSI EP17-A) and should be similar to that of the Old System. The specification criteria for the LoQ of the New System should be the same as for the Old System. We also recommend that the LoQ correspond to an analyte concentration level used in the precision studies.
We recommend that you conduct in-house within-laboratory precision studies (to supplement the external site reproducibility studies described below in Section c). When appropriate and justified, the in-house within-laboratory precision study may not be called for, for example, (i) if the manufacturer established that the New System only needs to be recalibrated at relatively long time intervals (e.g., 6 months or more) and any other concerns can be appropriately addressed by the reproducibility study, or (ii) if the New System is recalibrated daily, so that calibration cycle variability is inseparable from day-to-day variability (which is assessed by the reproducibility studies described below) and any other concerns can be appropriately addressed by the reproducibility study.
It may be sufficient to perform within-laboratory precision studies only on the New System. However, if the study design or composition of the precision panel of the Old System precision study was very different from that described in this guidance, it may be important to perform the precision study on the Old System as well. The within-laboratory precision study described below is based on modified CLSI document EP5-A2.
We recommend you evaluate samples with the following levels of analyte:
In addition, you should run the appropriate control material and calibrators associated with the test kit in the precision study.
If the assay has more than one medical decision point, then samples with concentrations around these medical decision points should be evaluated. It is understood that some assays will not have a specific medical decision point, but rather a range of values; in such cases, the panel should contain samples scattered throughout the measuring range of the assay.
Sources of variability we recommend for the within-laboratory precision study are at least 12 days of testing, with 2 runs per day, and 2 replicates of each sample per run. These 12 days are not necessarily consecutive and they should span at least two calibration cycles (the calibration cycles may be non-consecutive). For each cycle, you should include days at the beginning and end of the cycle (e.g., 3 days at the beginning and 3 days at the end of each cycle, for each cycle). You should include other additional sources of variability in the design of the study, if they are important to the specific assay (e.g., operators, lots, etc.). In such cases overall modification to the variables might be possible (e.g., spreading days of testing between different operators). If analytical and clinical performance is similar across all matrices that are included in the Intended Use of the Old System, then establishing performance of the New System using the most commonly employed matrix may suffice.We recommend you perform the reproducibility study based on a modification of CLSI EP15-A2 on the New System. The panel composition and analyte levels for this study should be the same as described in the within-laboratory precision study (Section B.1.b). We recommend that sources of variability should include testing for at least 5 days, 2 runs per day, with 3 replicates of each panel member per run at 3 laboratories (1 in-house and 2 external sites). Other sources of variability might be applicable if relevant to the specific assay (e.g., operators, etc.). If analytical and clinical performance is similar across all matrices that are indicated in the intended Use of the Old System, then establishing performance of the New System using the most commonly employed matrix may suffice.
For each concentration level, similar information should be available for the Old System. If this is not the case, a new reproducibility study for the Old System should be performed with study design and concentration levels as described in this section.
We recommend that you evaluate linearity for the New System according to CLSI EP6-A. The degree of linearity can be quantified using the maximum deviation from linearity (i.e., the delta described in CLSI document EP6-A). Your linearity study results should demonstrate that the delta of the New System is not greater than the observed delta in the linearity studies of the Old System. You should determine the appropriate number of replicates in the linearity study for the New System based on the precision studies of the New System.
You should perform comparison studies using comparison panels. Relatively minor changes to the Old System might not warrant all comparison studies. The extent of the utility of these studies can be evaluated on a case-by-case basis in consultation with the FDA.
For each analyte, the composition of the quantitative assay comparison panels should consist of at least 180 samples,8 including the following:
You should test the comparison panels on the Old System at a minimum of one site. This may be done in-house. However, you may want to use more than one Old System to better assess instrument bias. The New System should be tested at a minimum of three sites (one may be in-house) with at least one reagent kit lot. Each panel member should be tested at least four times: once with the Old System and three times with the New System. You should send the same positive and negative panels to each site, rather than dividing the panel between the three sites. Three different builds of the New System should be tested, one at each of the three sites.
Figure 4 Allowable Total Difference Zone
In addition to the acceptance criteria for LoB, LoD, LoQ and linearity, we recommend that you apply the following criteria:
Depending upon the unique characteristics of the qualitative or quantitative assay being migrated to the New System, the following studies may be called for. If not previously conducted for the Old System, they should be performed for the New System. If you determine that a study described below is not applicable for your system, you should describe your reasoning in detail in your application to FDA. FDA will consider such explanations on a case-by-case basis particularly for manual to semi-automated or automated System migrations.
There are specific criteria that are unique to nucleic acid tests (NAT) and therefore NATs present additional specific concerns over serological and antigen assays:
For the purposes of this document the following definitions are used. HTD referenced terms are based on the CLSI Harmonized Terminology Database.
Block diagrams : Engineering diagrams that graphically describe the instrument’s interior and exterior features, preferably to scale.
C5 : a concentration at which repeated tests of a sample with this concentration under stipulated conditions are 95% negative (or 5% positive) (see CLSI EP12-A2).
C95 : a concentration at which repeated tests of a sample with this concentration under stipulated conditions are 95% positive (see CLSI EP12-A2).
C50 :a concentration at which repeated tests of a sample with this concentration under stipulated conditions are 50 % positive (or 50% negative). Under ideal circumstances, C50 will exactly equal the cutoff established by the manufacturer.
Calibrators : a substance or device that is based on a reference preparation or in which the analyte concentration or other quantity has been determined by an analytical procedure of stated reliability. Calibrators are used to calibrate, graduate, or adjust a measurement [HTD].
Carry-over : amount of analyte carried by the measuring system from one sample reaction into subsequent sample reactions, thereby erroneously affecting the apparent amounts in subsequent samples [HTD].
Control material : a device, solution, or preparation intended for use in the quality control process to monitor the reliability of a test system and to maintain its performance within established limits.
Cross-reactivity : the ability of a drug, metabolite, a structurally similar compound other than the primary analyte, or even unrelated compound to affect the assay [HTD].
Cutoff value (CO) : for a qualitative test, the threshold above which the result of the test is reported as positive and below which the result is reported as negative. If a large series of tests were performed for a sample with concentration at the cutoff, 50% of test results will be positive and 50% will be negative; this analyte concentration can be termed C50 (CLSI EP12-A2).
High negative sample (C5) : a sample with a concentration of analyte close to the C5 as determined by the Old System . This term is equivalent to a “weak negative sample” for example as used in the CLIA Waiver Guidance document www.fda.gov/cdrh/oivd/guidance/1171.pdf
Hook effect (high dose hook effect) : effect caused by a decreasing signal response at very high levels of analyte. It is used interchangeably with “prozone effect,” the result of a suboptimal antigen-antibody reaction in which either the antibody or antigen is in excess resulting in an incomplete, or blocked reaction [HTD].
Interfering substances : endogenous (e.g., blood components, acidic polysaccharides) or exogenous (e.g., talc, anticoagulant) substances in clinical samples that can cause false-positive or false-negative results in a test system [HTD].
Limit of blank (LoB) : highest measurement result that is likely to be observed (with a stated probability) for a blank sample (a sample with concentration at or near zero) (CLSI EP17-A; [HTD]).
Limit of detection (LoD) : the lowest concentration of analyte that can be reported to be present at a specified level of confidence, although perhaps not quantified to an exact value. Similarly, an amount of analyte in a sample for which the probability of falsely claiming the absence is β (type II error) given a probability α (type I error) of falsely claiming its presence (CLSI EP17-A; [HTD])
Limit of quantification (LoQ) : the lowest amount of analyte in a sample that can be quantitatively determined with {stated} acceptable precision and {stated, acceptable} accuracy, under stated experimental conditions (CLSI EP17-A; [HTD]).
Linearity studies : studies to determine the analyte concentration range over which the testing systems results are acceptably linear with the ability (within a given range) to provide results that are directly proportional to the concentration (amount) of the analyte in the test sample.
Low positive sample (C95) : a sample with a concentration of analyte close to C 95 as determined by the Old System . This term is equivalent to “weak positive sample” for example as used in the CLIA Waiver Guidance document, www.fda.gov/cdrh/oivd/guidance/1171.pdf.
Measurand : particular quantity subject to measurement. The term “measurand” and its definition encompass all quantities while the commonly used term “analyte” refers to a tangible entity subject to measurement [HTD].
Measuring range : set of values of measurands for which the error of a measuring instrument is intended to lie within specified limits. The range of values (in units appropriate for the analyte [measurand]) over which the acceptability criteria for the method have been met; that is, where errors due to nonlinearity, imprecision, or other sources are within defined limits (CLSI EP6-A, [HTD]).
Medical decision level (medical decision point) : a level or concentration at which a test is critically interpreted for patient care and treatment.
Moderate positive sample : a sample with a concentration close enough to the cutoff and at which one can anticipate positive results by the Old System approximately 100% of the time.
Negative percent agreement : the proportion of samples negative by the Old System for which the results by the New System are negative (see FDA guidance at www.fda.gov/cdrh/osb/guidance/1620.pdf).10
New System : an unapproved/unlicensed system (assay, instrument, and software) to which the assay is migrating from a previously approved/licensed system.
Old System : an approved/licensed system (assay, instrument and software) from which the assay is migrating to a currently unapproved/unlicensed system.
Positive percent agreement : the proportion of samples positive by the Old System for which the results by the New System are positive (see FDA guidance at www.fda.gov/cdrh/osb/guidance/1620.pdf) .
Repeatability : closeness of the agreement between the results of successive measurements of the same measurand carried out under the same conditions of measurement (See CLSI EP5-A2, [HTD]).
Reproducibility : closeness of agreement between the results of measurements of the same measurand and carried out under changed conditions of measurement. Reproducibility conditions are conditions where test results are obtained with the same method on identical test items in different laboratories with different operators using different equipment and can include additional variables such as days, replicates, and runs (See CLSI EP5-A2, [HTD])
Risk analysis : systematic use of available information to identify hazards and to estimate the risk. Risk analysis includes examination of different sequences of events that can produce hazardous situations and harm [HTD].
Spiked sample : a clinical sample to which has been added exogenous analyte to create specified levels of signal.
Systematic difference : a mean of the measurand on the New System minus a value of the same measurand as performed on the Old System that would result from an infinite number of measurements carried out under the stipulated condition (based on HTD).
Within-laboratory precision: precision over a defined time and operators, within the same facility and using the same equipment; calibration and reagents may vary. Formerly, the term “ total precision” was used in CLSI EP5-A2 [HTD] .
CDRH Guidance Documents:
Clinical and Laboratory Standards Institute (CLSI) documents:
Blood Donor Screening Assays for infectious agents, reviewed under Biologics License Applications, are generally held to stringent standards of sensitivity and specificity. Typically, clinical studies for licensure of products such as HIV assays involve testing of over 1000 known positives and 6000 to 10,000 low risk samples (or pools) collected from the intended use population. Consequently, FDA recommends larger study sizes for migrating blood screening assays to New Systems.
Otherwise, except as specifically noted below, the same considerations apply to blood screening assays as described for qualitative assays in Section VI.A of this document, “Migration Studies for Qualitative Assays.”
For immunoassays, FDA recommends that at least 20 seroconversion panels, or as many as are available (whichever is less) be studied, comparing the New and the Old Systems head to head. For nucleic acid tests (NAT), FDA recommends the head-to-head testing of as many seroconversion panels as were tested for licensure of the Old System (typically 10). Both qualitative results and S/CO ratios should be compared.
FDA recommends that sponsors compare the New and Old Systems in a Precision Study as outlined in Section VI.A.1.b
FDA recommends that sponsors compare the New and Old Systems in a Reproducibility Study essentially as outlined in Section VI.A.1.c. However, FDA recommends including in the panel at least one truly negative sample (other than a positive sample diluted to below the cutoff), and that testing be performed at three sites, of which one may be in-house.
Option 1: A positive comparison panel should consist of approximately 100 positive samples, to include 20-30 specimens with signals ≤3X the cutoff for an immunoassay or analyte concentrations ≤3X the 95% LoD for a NAT. Specimens may be diluted to this range if not clinically available. The panel should be tested on the New System at three sites, one of which can be in-house. This panel should also be tested at least once on the Old System (this can be in-house). The data should be analyzed by S/CO regression and analysis of bias using scatter-plots or similar graphical presentations, as described in Section VI.A.3.c.
Option 2: As an alternative option, the sponsor may wish to test head to head on the New and Old Systems at three external sites the lowest 10% of positive specimens from the original clinical trial of the assay on the Old System, if the specimens have been stored under conditions defined in the instructions for use of the assay.
Option 1: A negative comparison panel should consist of 3000 known negative samples (or pools), or specimens obtained from a low risk study, with adequate follow-up. The panel should be tested on the New System, distributed over three sites, one of which may be in-house (e.g., 1/3 at site one, 1/3 at site two & 1/3 at site three). The data should be analyzed for agreement of the point estimate of specificity (with the 95% confidence interval) for the New System with the original point estimate of specificity (with the 95% confidence interval) from the original trial of the Old System.
Option 2: As an alternative option, the sponsor may wish to test head to head on the Old and New Systems at three external sites the highest 10% of negative specimens from the original clinical trial of the assay on the Old System, if the specimens have been stored under conditions defined in the instructions for use of the assay.
Except as noted above, the same acceptance criteria as recommended in Section VI.A.4 also apply to blood donor screening assays.
Additionally, sponsors may recommend statistical analysis protocols based on estimating false positive and false negative ratios from the negative and positive comparison panel studies, respectively. Sponsors interested in this approach should determine an appropriate model for the S/CO distribution of each panel (positive or negative) together with a proposed method of analysis.
Only substances and conditions that represent a reasonable risk of interference in the New System should be studied. For instance, interfering conditions such as hemolysis or hyperlipidemia might influence pipetting or washing steps and should be included in migration studies. Conversely, it would seem unlikely that cross-reactivity of, for instance, an HIV NAT assay with HTLV would likely be influenced by migration to a New System.
Each interfering substance/condition may be tested in-house using a panel of approximately 10 low positives with signals <3X the cutoff for an immunoassay or analyte concentrations <3X the 95% LoD for a NAT.
Each interfering substance/condition may be tested in-house using a panel of approximately 10 true negatives.
Consider that an assay (qualitative or quantitative) has a numerical output. If the standard deviations (SD) in the precision studies of the Old System for concentrations around the cutoff value are almost constant, then:
C95 = C50 +1.645 x SD, and
C5 = C50 – 1.645 x SD
For example, if the cutoff for optical density (OD) value is 1.00 and the SD around the cutoff is approximately 0.10, then C95 is approximately 1.16 OD (=1.00+1.645 x 0.10) and C5 is approximately 0.84 OD (=1.00 -1.645 x 0.10). In other words, a sample with an actual OD of 1.16 produces positive results (above 1.00) approximately 95% of the time and a sample with an actual OD of 0.84 produces negative results (below 1.00) approximately 95% of the time.
If the coefficient of variation (CV) in the precision studies of the Old System for concentrations around the cutoff value are almost constant, then
C95 = C50 + 1.645 x CV x C95 and C5 = C50 – 1.645 x CV x C5. From here,
C95 = C50 / (1 – 1.645 x CV) and
C5 = C50 / (1 + 1.645 x CV).
For example, if the cutoff has an OD value of 1.00 and the %CV around the cutoff is approximately 10% (i.e., CV=0.10), then C95 is approximately 1.20 OD and C5 is approximately 0.86 OD.
If the limit of blank (LoB) is used as a cutoff, then the concentration C95 is the same as the limit of detection (LoD) and zero concentration is C5 (see CLSI EP17-A).
Positive panel samples:
|
Old System Positive |
||
|---|---|---|---|
|
Low Positive |
Moderate Positive
|
High Positive
|
New System Positive |
27 |
30 |
40 |
New System Negative |
3 |
|
|
Total |
30 |
30 |
40 |
Negative panel samples:
|
Old System Negative |
|
|---|---|---|
|
Low and Moderate Negative
|
High Negative |
New System Positive |
|
3 |
New System Negative |
70 |
27 |
Total |
70 |
30 |
If the CO of the assay is the LoB, the columns of Low Negative and Moderate Negative can be combined as in the example above.
The following are additional recommendations for performing statistical analyses of percentages or proportions. Confidence limits for positive percent agreement and negative percent agreement can be calculated using formulas for calculating a confidence interval for a binomial proportion. There are several different methods available. We suggest that either a score method described by Altman, et al. (Altman D.A., Machin D., Bryant T.N., Gardner M.J. eds. Statistics with Confidence. 2 nd ed. British Medical Journal; 2000) or a Clopper-Pearson Method (Clopper CJ, Pearson E. Biometrika 1934; 26:404-413) be used.
An advantage with the score method is that it has better statistical properties and it can be calculated directly. Score confidence bounds tend to yield narrower confidence intervals than Clopper-Pearson confidence intervals, resulting in a larger lower confidence bound. So with n=100 samples and 96/100=96% agreement, the score lower confidence bound is 90.2% whereas the Clopper-Pearson lower confidence bound is 90.1%. In this document, we have illustrated the reporting of confidence intervals using the score approach. For convenience, we provide the formulas for the score confidence interval for a percentage. Note that the lower bound of a two-sided 95% score confidence interval is the same as the lower bound of a one-sided 97.5% score confidence interval; and the lower bound of one-sided 95% score confidence interval is the same as the lower bound of a two-sided 90% score confidence interval.
A two-sided 95% score confidence interval for the proportion of A/B is calculated as:
[100%(Q1 - Q2) / Q3, 100%(Q1 + Q2) / Q3], where the quantities Q1, Q2, and Q3 are computed from the data using the formulas below. For the proportion of A/B:
Q1 = 2 · A + 1.962 = 2 · A + 3.84
Q2 = 1.96√(1.962 + 4 · A · (B - A) / B) = 1.96√(3.84 + 4 · A · (B - A) / B)
Q3 = 2 · (B + 1.962) = 2 · B + 7.68
In the formulas above, 1.96 is the quantile from the standard normal distribution that corresponds to 95% confidence. For calculation of 95% one-sided score confidence interval, use 1.645 in place of 1.96 in the formulas above.
| Observed Difference From Clinical Point of View | Observed Difference From Statistical Point of View | Interpretation |
|---|---|---|
| Small | Non-significant* | Acceptable |
| Small | Significant* | Acceptable |
| Large | Non-Significant | Larger sample size is likely needed |
| Large | Significant | Not acceptable |
* Confidence interval is within clinically acceptable differences
For a panel of 100 samples which test positive by the Old System, and of which 96 also test positive by the New System (96 out of 100), the lower limit of the 95% two-sided score confidence interval is above 90%. For 30 samples with values close to C95, the 95% two-sided confidence interval for 26/30 (87%) is 70.3% to 94.7%. If, for example, among the 30 samples with low positive concentrations (concentrations close to C95 by the Old System), only 25 samples test positive by the New System, then the percent of positive results by the New System for the samples close to the cutoff is statistically different from 95% (83% (25/30) with 95% CI: 66.4% to 92.7%).
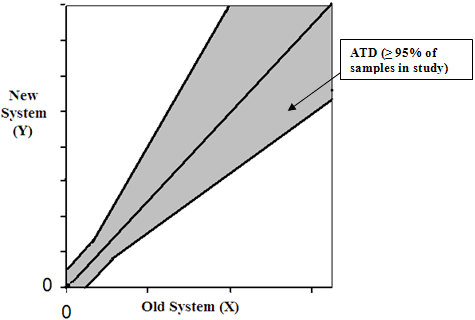
For each sample of the Comparison panel, calculate the differences between the New System result (Y) and the Old System result (X), Y-X (based on CLSI EP21). Also calculate (X+Y)/2. Plot the difference between Y and X, Y-X, against their mean (Y+X)/2 (Bland-Altman plot). On the Bland-Altman plot of (Y-X) vs. (Y+X)/2, provide the Allowable Total Difference (ATD) zone around the axis, (Y+X)/2. The ATD zone is established in such a way that 95% of differences between the Old System result and the repeated result by the Old System fall within the ATD. The ATD zone is expressed as:
± 1.96 · √2 · CV· (Y+X)/2 = ± 2.77· CV· (Y+X)/2 for larger values of Old System and ± 1.96 ·√2 · SD = ± 2.77· SD for the low values of Old System where CV and SD are the reproducibility characteristics of the Old System (see Establishing SD and percent CV for ATD Based on the Performance of the Old System below). A hypothetical example of the ATD zone on the Bland-Altman plot is provided below (Figure 5):
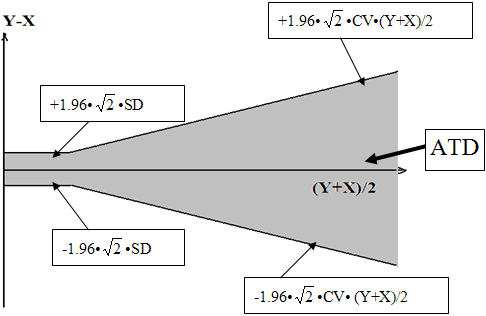
Figure 5. A hypothetical example of the ATD zone on the Bland-Altman plot is provided below.
By the appropriate transformation, a similar ATD zone can be presented on the plane of New System values (Y) against Old System values (X), see Figure 4. The expressions of the lines of the ATD zone on the plane Y vs. X are the following:
Lines parallel to the diagonal of the ATD zone:
Y = X ± 1.96 · √2 · SD = X ± 2.77 · SD, if 0 ≤ Y + X ≤ 2 · A
Lines for the “expanding” part of the ATD zone are:
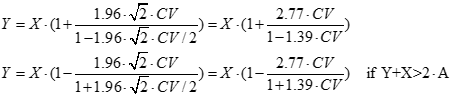
where A is SD/CV.
For an individual measurement Xi of a given sample by the Old System, there is a following expression: Xi = Xtruei + Mean-Bias + Random-Biasi + εi where deviation of Xi from the true value Xtruei is composed of a mean bias, a random matrix-related interference component, and a random measurement error12,13. Because there are no changes in the assay, it is anticipated that the random matrix- related interferences in both systems are the same. Then the difference between New System and Old System measurements of the same sample depends on a random measurement error.
Establishing standard deviation (SD) for the Allowable Total Difference (ATD) zone should be based on consideration of possible variance between the two measurements of the same sample by the Old System obtained at different sites. For each concentration in the precision study, the largest SD among three sites in the precision study may be selected with addition of the between-site component of variance. For example, if for some concentration X1, three sites have SD of precision as 0.111 (site 1), 0.086 (site 2) and 0.118 (site 3) with between-site component of 0.020; then the standard deviation of two measurements by Old System performed in two different sites similar to site 3 (site with highest imprecision) is sqrt(0.118*0.118+0.020*0.020)= 0.120.
Another hypothetical precision experiment for the Old System can produce a slightly higher SD than in this example. In order to address this, the observed SD should be multiplied by the appropriate factor (factor = (1 - 1 / (4 · f)-1) · √(f / x25%(f)), f is a degree of freedom of the estimated SD).
For example, if the degrees of freedom of the SD in the precision study of the Old System was 40, then the appropriate factor is 1.236 and the expected maximum observed SD can be as high as 0.148 (=0.120 *1.236). After the appropriate SD or percent CV is established for each concentration in the precision study, the ATD zone can be obtained by smooth interpolation.
For 150 samples with 95% of the observations (143 /150) falling in the ATD zone, the lower limit of the 95% one-sided confidence interval is above 90%.
1 This guidance does not apply to immunohematology tests licensed by the Center for Biologics Evaluation and Research (CBER).
2 This guidance can be used for 510(k) devices where the Replacement Reagent and Instrument Family Policy (http://www.fda.gov/cdrh/oivd/guidance/950.pdf) does not apply (e.g., nucleic acid amplification tests) and devices for which transition to a New System presents specific concerns, either because of the nature of the analyte and indications, or because of the specific technology used.
3 http://www.fda.gov/cdrh/oivd/guidance/950.pdf.
4 http://www.fda.gov/oc/initiatives/criticalpath/whitepaper.html
5 FDA does not believe that this guidance is suitable for use in its entirety when immunohematology tests (e.g., blood grouping, blood group antibody detection and/or identification, crossmatching) are being migrated because of the differences in assay methodology and results reading and interpretation as compared to the other assays and systems described in this guidance. If you believe that your immunohematology reagents and system can be evaluated using the criteria outlined in this guidance, contact the responsible review division in CBER. Immunohematology products are reviewed in the Devices Review Branch, Division of Blood Applications, Office of Blood Research and Review, CBER.
6 ISO 14971:2007, Medical devices – Application of risk management to medical devices, and Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices.
7 Guidance for Industry and FDA Staff: Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices, http://www.fda.gov/cdrh/ode/guidance/337.html.
8 For more information on sample size, please refer to Appendix II: Statistical Notes, 7.
9 Commission Decision 2002/364/EC of 7 May 2002 on common technical specifications for in vitro-diagnostic medical devices [Official Journal L 131 of 16.05.2002].
10 The general definition from the cited guidance is adapted in this guidance since the cited guidance refers to clinical subjects, whereas this guidance does not involve subjects, but rather specimens or samples. In addition, the term “non-reference standard” in the cited guidance is analogous to “Old System” in this guidance; the term “test” in the cited guidance is analogous to “New System”.
11 Appendix 1 does not apply to immunohematology tests licensed by CBER.
12 Krouwer, JS. Estimating total analytical error and its sources. Techniques to improve method evaluation. Arch. Pathol. Lab. Med. 1992; 116:726-731.
13 Linnet K. Boyd JC. Analytical validation of methods – With statistical methods. In: Burtis C, Ashwood ER, Bruns D (eds) Tietz Textbook of Clinical Chemistry and Molecular Diagnostics. 4 ed. New York: Saunders, 2006, p.353-407
Updated January 2, 2009
![]()
CDRH Home Page | CDRH A-Z Index | Contact CDRH | Accessibility | Disclaimer
FDA Home Page | Search FDA Site | FDA A-Z Index | Contact FDA | HHS Home Page
Center for Devices and Radiological Health / CDRH



