
U.S. Department of Health and Human Services
|
 |
Thanks to the following organizations for their invaluable assistance in preparing this report:
OIVD Management Staff
OIVD Review Divisions
ODE Program Operations Staff
OMO Division of Planning, Analysis and Finance
OMO Division of Information Technology Management
Kathy Hobbs, ODE Editor
MaryAnn Gornick, ODE Production SpecialistDonald St.Pierre, Deputy Director
Mary Pastel, Project Director
Patricia Beverly, Project Manager
Eugene Reilly, Reviewer
The Office of In Vitro Diagnostic Device Evaluation and Safety (OIVD) is now 3 years old. We remain firmly committed to the objective put on the table when our organization was first launched: to actualize total product life cycle regulation for In Vitro Diagnostic Devices (IVDs).
During 2005 we have made significant strides in meeting that goal. We have completed processes to integrate all three critical regulatory functions for IVDs - premarket review, compliance oversight, and postmarket surveillance - into a single functional unit.
While we have worked studiously to ensure our activities are congruent with work throughout our center, our work differs in several ways. It is now driven from a common technical base which allows both external and internal stakeholders one stop shopping for IVD information. In addition, we have begun to utilize the geographic and organizational features of our office to create powerful and growing connections between our disparate regulatory programs.
This integration of diverse work has been accomplished while meeting the challenges of our routine work and includes: 11 premarket approval application (PMA) reviews, 520 premarket notification 510(k) reviews, 7 Class I recalls, 3 warning letters and 9605 MDR reviews; implementing MDUFMA hiring, training and goals; and identifying and training staff to perform new regulatory functions in a multifunctioning, matrix driven organization.
While we believe we have made a significant start toward creating a unique new office with its own identity, culture, and work processes, our eye is on the future. The IVD industry is brimming with new ideas, technologies, and biological insights that are likely to create a diagnostic revolution in medicine in the next 5 to 10 years. In particular, the drive toward personalized medicine and the use of diagnostics to shorten the critical path to introduction of new drugs, the interest in diagnostic markers for bioterrorism threat preparedness and for dealing with emerging infectious disease such as avian flu, and the development of portable systems to increase access to rapid testing, are likely to drastically impact patient health and safety.
OIVD is up for the challenge of becoming an engaged and proactive member of this diagnostic revolution. It is our hope that our role at the table will be one of facilitator and partner rather than obstacle. We are committed to the important mission of promoting and maintaining availability of safe and effective diagnostics for use in the medical marketplace.
Steven Gutman, M.D., M.B.A.
Director
Office of In Vitro Diagnostic Device
Evaluation and Safety
We work to promote and protect public health through clear and consistent regulation of in vitro diagnostic devices (IVDs). We have a dual charge to foster the rapid transfer of good new IVDs into the medical market while preventing marketing of unsafe or ineffective devices. We strive to ensure our work is transparent and allows all stakeholders to obtain the knowledge required to make informed decisions about the development, production, and use of IVDs.
OIVD provides timely, comprehensive, and integrated regulatory oversight for IVDs through our Total Product Life Cycle (TPLC) program by:
Last year, the Office of In Vitro Diagnostic Device Evaluation and Safety (OIVD) approved and cleared hundreds of devices used as an aid in diagnosis. Below, we highlight several new medical devices and devices with new indications approved or cleared during this past fiscal year that we believe will have a particular impact on patient care.
Cleared December 13, 2004, Vysis® AutoVysion™ System, manufactured by Vysis, Inc., is a device that consists of an automated scanning microscope, image analysis system and customized software applications for fluorescence in situ hybridization (FISH) assays. This device is intended for in vitro diagnostic use with FISH assays as an aid in the detection, counting and classification of cells based on recognition of cellular color, size and shape and in the detection and enumeration of FISH signals in interphase nuclei of formalin-fixed, paraffin-embedded human tissue specimens. Link to clearance: http://www.fda.gov/cdrh/mda/docs/k041875.html
 Approved December 22, 2004, ADVIA Centaur® HCV, manufactured by Bayer Healthcare, LLC, is an in vitro diagnostic immunoassay for the qualitative determination of immunoglobulin G (IgG) antibodies to hepatitis C virus (HCV) in human serum and plasma (EDTA, lithium or sodium heparinized) using the ADVIA Centaur® System. The assay may be used in conjunction with other serological and clinical information to aid in the diagnosis of individuals with symptoms of hepatitis and in individuals at risk for hepatitis C infection. ADVIA HCV Quality Control Materials: For in vitro diagnostic use in monitoring the performance of the HCV assay on the ADVIA Centaur® Systems. The performance of the HCV quality control material has not been established with any other anti-HCV assays. Link to approval: http://www.fda.gov/cdrh/mda/docs/p030056.html
Approved December 22, 2004, ADVIA Centaur® HCV, manufactured by Bayer Healthcare, LLC, is an in vitro diagnostic immunoassay for the qualitative determination of immunoglobulin G (IgG) antibodies to hepatitis C virus (HCV) in human serum and plasma (EDTA, lithium or sodium heparinized) using the ADVIA Centaur® System. The assay may be used in conjunction with other serological and clinical information to aid in the diagnosis of individuals with symptoms of hepatitis and in individuals at risk for hepatitis C infection. ADVIA HCV Quality Control Materials: For in vitro diagnostic use in monitoring the performance of the HCV assay on the ADVIA Centaur® Systems. The performance of the HCV quality control material has not been established with any other anti-HCV assays. Link to approval: http://www.fda.gov/cdrh/mda/docs/p030056.html
Approved December 22, 2004, ADVIA Centaur® HBc, manufactured by Bayer Healthcare, LLC, is an in vitro diagnostic test for the qualitative determination of total antibodies to the core antigen of the hepatitis B virus (HBc Total) in human serum or plasma (potassium EDTA, or lithium or sodium heparinized) using the ADVIA Centaur® System. This assay can be used as an aid in the diagnosis of individuals with acute or chronic hepatitis B virus (HBV) infection and in the determination of the clinical status of HBV-infected individuals in conjunction with other HBV serological markers for the laboratory diagnosis of HBV disease associated with HBV infection. This assay can also be used as an aid in the differential diagnosis in individuals displaying signs and symptoms of hepatitis in whom etiology is unknown. HBc Total Quality Control Materials: The controls are indicated for in vitro diagnostic use in monitoring the performance of the HBc Total assay on the ADVIA Centaur® Systems. The performance of the HBc Total Quality Control Material has not been established with any other anti-HBc Total assays. Link to approval: http://www.fda.gov/cdrh/mda/docs/p040004.html
Approved December 22, 2004, ADVIA Centaur® HAV IgM, manufactured by Bayer Healthcare, LLC, is indicated for use for the ADVIA Centaur® HAV IgM assay and is an in vitro diagnostic immunoassay for the qualitative determination of IgM response to the hepatitis A virus (HAV) in human serum or plasma (potassium EDTA, lithium or sodium heparinized) using the ADVIA Centaur® System. The assay is intended for use as an aid in the diagnosis of acute or recent infection (usually 6 months or less) with hepatitis A virus. HAV IgM Quality Control Materials: The controls are indicated for in vitro diagnostic use in monitoring the performance of the HAV IgM assay on the ADVIA Centaur® Systems. The performance of the HAV IgM quality control material has not been established with any other anti-HAV IgM assays. Link to approval: http://www.fda.gov/cdrh/mda/docs/p040018.html
 Cleared December 23, 2004, the Affymetrix GeneChip® Microarray Instrumentation System consisting of GeneChip® 3000Dx scanner with autoloader, FS450Dx fluidics station and GCOSDx software is intended to measure fluorescence signals of labeled DNA target hybridized to GeneChip® arrays. It was cleared for use with the AmpliChip CYP450 Test, manufactured by Roche Molecular Systems as drug metabolizing enzyme genotyping system. Link to clearance: http://www.fda.gov/cdrh/pdf4/k042279.pdf
Cleared December 23, 2004, the Affymetrix GeneChip® Microarray Instrumentation System consisting of GeneChip® 3000Dx scanner with autoloader, FS450Dx fluidics station and GCOSDx software is intended to measure fluorescence signals of labeled DNA target hybridized to GeneChip® arrays. It was cleared for use with the AmpliChip CYP450 Test, manufactured by Roche Molecular Systems as drug metabolizing enzyme genotyping system. Link to clearance: http://www.fda.gov/cdrh/pdf4/k042279.pdf
 Cleared December 23, 2004, the AmpliChip CYP450 Test, manufactured by Roche Molecular Systems, is a drug metabolizing enzyme genotyping system. It is the first microarray cleared by FDA for clinical use and is intended for use in testing DNA to identify the presence or absence of human genotypic markers encoding two drug metabolizing enzymes. The device is used as an aid in determining treatment choice and individualizing treatment dose for therapeutics that are metabolized primarily by the specific enzyme about which the system provides genotypic information. Link to clearance: http://www.fda.gov/cdrh/mda/docs/k042259.html
Cleared December 23, 2004, the AmpliChip CYP450 Test, manufactured by Roche Molecular Systems, is a drug metabolizing enzyme genotyping system. It is the first microarray cleared by FDA for clinical use and is intended for use in testing DNA to identify the presence or absence of human genotypic markers encoding two drug metabolizing enzymes. The device is used as an aid in determining treatment choice and individualizing treatment dose for therapeutics that are metabolized primarily by the specific enzyme about which the system provides genotypic information. Link to clearance: http://www.fda.gov/cdrh/mda/docs/k042259.html
Approved January 24, 2005, UroVysion™ Bladder Cancer Kit, manufactured by Vysis, Inc., is designed to detect aneuploidy for chromosomes 3, 7, 17 and loss of the 9p21 locus via fluorescence in situ hybridization (FISH) in urine specimens from persons with hematuria suspected of having bladder cancer. Results from the UroVysion Kit are intended for use in conjunction with and not in lieu of current standard diagnostic procedures, as an aid for initial diagnosis of bladder carcinoma in patients with hematuria and subsequent monitoring for tumor recurrence in patients previously diagnosed with bladder cancer. Link to approval: http://www.fda.gov/cdrh/mda/docs/p030052.html
Approved March 7, 2005, ADVIA Centaur® HAV, manufactured by Bayer Healthcare, LLC, is an in vitro diagnostic immunoassay for the qualitative determination of total antibodies to hepatitis A virus (anti-HAV ) in human serum or plasma (potassium EDTA, lithium or sodium heparinized) using the ADVIA Centaur® System. This anti-HAV assay is indicated as an aid in the diagnosis of previous or ongoing hepatitis A viral infection or in the identification of HAV-susceptible individuals for vaccination. Link to approval: http://www.fda.gov/cdrh/mda/docs/p040017.html
Cleared April 18, 2005, PAXgene™ Blood RNA, manufactured by PreAnalytiX GMBH, is intended to collect, store, and transport patient specimens and stabilize intracellular RNA from the specimens for subsequent isolation and purification of the intracellular RNA for RT-PCR used in vitro molecular diagnostic testing. Link to clearance: http://www.fda.gov/cdrh/mda/docs/k042613.html
 Approved May 3, 2005, Her2 FISH pharmDx™ Kit, manufactured by DakoCytomation Denmark A/S, is a direct fluorescence in situ hybridization (FISH) assay designed to quantitatively determine the Her2 gene amplification in formalin-fixed, paraffin-embedded breast cancer tissue specimens. Her2 FISH pharmDx™ Kit is indicated as an aid in the assessment of patients for whom Herceptin® (trastuzumab) treatment is being considered. Results form the Her2 FISH pharmDx™ Kit are intended for use as an adjunct to the clinicopathologic information currently used for estimating prognosis in stage II, node positive breast cancer patients. Link to approval: http://www.fda.gov/cdrh/mda/docs/p040005.html
Approved May 3, 2005, Her2 FISH pharmDx™ Kit, manufactured by DakoCytomation Denmark A/S, is a direct fluorescence in situ hybridization (FISH) assay designed to quantitatively determine the Her2 gene amplification in formalin-fixed, paraffin-embedded breast cancer tissue specimens. Her2 FISH pharmDx™ Kit is indicated as an aid in the assessment of patients for whom Herceptin® (trastuzumab) treatment is being considered. Results form the Her2 FISH pharmDx™ Kit are intended for use as an adjunct to the clinicopathologic information currently used for estimating prognosis in stage II, node positive breast cancer patients. Link to approval: http://www.fda.gov/cdrh/mda/docs/p040005.html
 Cleared May 9, 2005, Tag-It™ Cystic Fibrosis Kit, manufactured by Tm BioScience Corp., is a device used to simultaneously detect and identify a panel of mutations and variants in the CFTR gene. It is intended as an aid in confirmatory diagnostic testing of individuals with suspected cystic fibrosis (CF), carrier identification and newborn screening. This device is not intended for stand-alone diagnostic purposes, prenatal diagnostic, pre-implantation or population screening. Link to clearance: http://www.fda.gov/cdrh/mda/docs/k043011.html
Cleared May 9, 2005, Tag-It™ Cystic Fibrosis Kit, manufactured by Tm BioScience Corp., is a device used to simultaneously detect and identify a panel of mutations and variants in the CFTR gene. It is intended as an aid in confirmatory diagnostic testing of individuals with suspected cystic fibrosis (CF), carrier identification and newborn screening. This device is not intended for stand-alone diagnostic purposes, prenatal diagnostic, pre-implantation or population screening. Link to clearance: http://www.fda.gov/cdrh/mda/docs/k043011.html
 Cleared May 19, 2005, Wako LBA Alpha Fetoprotein (AFP)-L3, AFP-L3 Set, manufactured by Wako Chemicals USA, Inc., is an in vitro diagnostic device that consists of reagents and an automated instrument used to quantitatively measure AFP and AFP-L3 subfraction in human serum by immunochemical techniques. The device is intended for in vitro diagnostic use as an aid in the risk assessment of patients with chronic liver disease for progression to hepatocellular carcinoma in conjunction with other laboratory findings, imaging studies and clinical assessment. Link to clearance: http://www.fda.gov/cdrh/mda/docs/k041847.html
Cleared May 19, 2005, Wako LBA Alpha Fetoprotein (AFP)-L3, AFP-L3 Set, manufactured by Wako Chemicals USA, Inc., is an in vitro diagnostic device that consists of reagents and an automated instrument used to quantitatively measure AFP and AFP-L3 subfraction in human serum by immunochemical techniques. The device is intended for in vitro diagnostic use as an aid in the risk assessment of patients with chronic liver disease for progression to hepatocellular carcinoma in conjunction with other laboratory findings, imaging studies and clinical assessment. Link to clearance: http://www.fda.gov/cdrh/mda/docs/k041847.html
Approved May 26, 2005, ADVIA Centaur® HBsAg, manufactured by Bayer Healthcare, LLC, is an in vitro immunoassay for the qualitative detection of hepatitis B surface antigen (HBsAg) in human serum and plasma (potassium EDTA, lithium or sodium heparinized) using the ADVIA Centaur® system. The assay may be used in conjunction with other serological and clinical information to diagnose individuals with acute or chronic hepatitis B infections. The assay may also be used to screen for hepatitis B infection in pregnant women to identify neonates who are at risk of acquiring hepatitis B during the perinatal period. ADVIA Centaur HBsAg Confirmatory Ready Pack: The ADVIA Centaur® HBsAg Confirmatory assay is an in vitro diagnostic immunoassay for the qualitative confirmation of the presence of hepatitis B surface antigen (HBsAg) in human serum or plasma (EDTA, lithium or sodium heparin) using the ADVIA Centaur® system. The assay is intended to be used to confirm the presence of HBsAg in samples that are repeatedly reactive using the ADVIA Centaur HBsAg assay. ADVIA Centaur® HBsAg Quality Control Material Indications for Use: for monitoring the performance of the HBsAg and HBsAg Confirmatory assays on the ADVIA Centaur® Systems. Link to approval http://www.fda.gov/cdrh/MDA/DOCS/p030049.html
Approved June 27, 2005, c-Kit pharmDx™, manufactured by DakoCytomation Denmark A/S, is a qualitative immunohistochemical (IHC) kit system used on the Dako Autostainer, for the identification of c-kit protein/CD117 antigen (c-kit protein) expression in normal and neoplastic formalin-fixed paraffin-embedded tissues for histological evaluation. The c-Kit pharmDx™ rabbit polyclonal antibodies specifically detect the c-kit protein in CD117 antigen-expressing cells. The c-Kit pharmDx™ is indicated as an aid in the differential diagnosis of gastrointestinal stromal tumors (GIST). After diagnosis of GIST, results from c-Kit pharmDx™ may be used as an aid in identifying those patients eligible for treatment with Gleevec™/Glivec® (imatinib mesylate). Results from hematoxylin and eosin (H&E) stains and a panel of antibodies can aid in the differential diagnosis of GIST. Interpretation must be made by a qualified pathologist, within the context of a patient’s clinical history, proper controls, and other diagnostic tests. Link to approval: http://www.fda.gov/cdrh/mda/docs/p040011.html
Cleared July 6, 2005, Joint Biological Agent Identification and Diagnostic System (JBAIDS) Anthrax Detection System, manufactured by Idaho Technology, Inc. The JBAIDS Anthrax Target 1 Assay is a qualitative IVD test for the detection of 1 of 2 DNA sequence targets, both of which are essential for the organism’s pathogenicity. The JBAIDS Anthrax Target 2 Assay is a qualitative IVD test for the detection of the second DNA sequence target, and is run as a confirmatory test after obtaining a Positive result from the Target 1 Assay. The results from these tests are used in conjunction with culture and other laboratory test and clinical information as an aid in the diagnosis of systemic anthrax infection in individuals suspected of having the disease. Link to clearance: http://www.fda.gov/cdrh/pdf5/k051713.pdf
Approved July 18, 2005, Guardian® RT, manufactured by Medtronic MiniMed, is indicated for continuous or periodic monitoring of glucose levels in the fluid under the skin in adults (ages 18 and older) with diabetes mellitus for the purpose of improving diabetes management. It alerts if a glucose level falls below or rises above preset values. Values are not intended to be used directly for making therapy adjustments, but rather to provide an indication of when a finger stick may be required. All therapy adjustments would be based on measurements obtained using a home glucose monitor and not on Guardian values. Guardian® RT provides real-time glucose values that allow users to track patterns in glucose concentrations and to possibly identify episodes of low and high blood glucose episodes. It also stores the data so that it can be analyzed to track patterns. Glucose data can be further downloaded to PC software for analysis of historical glucose values. Link to approval: http://www.fda.gov/cdrh/pdf/p980022s011.html
Cleared August 16, 2005, Gamma Phage Lysis Assay, manufactured by the Office of the Surgeon General, is a lytic phage assay specific for Bacillus anthracis and may be used as an aid for identification of Bacillus anthracis. The Gamma Phage assay can be used on suspect non-hemolytic, aerobic, gram-positive, “ground-glass” appearing colonies from sheep blood agar in conjunction with other markers and testing for the identification of Bacillus anthracis. This assay is not intended for screening of blood or plasma donors. Link to clearance: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMN/pmn.cfm
Cleared August 22, 2005, the Invader® UGT1A1 Molecular Assay, manufactured by Third Wave Technologies, Inc., is a new blood test that will help doctors make personalized drug treatment decisions for some patients. The Invader® UGT1A1 Molecular Assay detects variations in a gene that affects how certain drugs are broken down and cleared by the body. Doctors can use this information to help determine the right drug dosage for individual patients, and minimize harmful drug reactions. The Invader assay is intended to aid a physician in making individualized patient treatment decisions. Link to clearance: http://www.fda.gov/cdrh/pdf5/k051824.pdf
| Document # | Date | Applicant | Device |
|---|---|---|---|
| P040030 | 12/17/04 | BioGenex Laboratories, Inc. | InSite™ Her-2/neu Kit (withdrawn after approval) |
| P030056 | 12/22/04 | Bayer Healthcare, LLC | ADVIA Centaur® HCV |
| P040004 | 12/22/04 | Bayer Healthcare, LLC | ADVIA Centaur® HBc Total ReadyPack Reagents |
| P040018 | 12/22/04 | Bayer Healthcare, LLC | ADVIA Centaur® HAV IgM ReadyPack Reagents |
| P030052 | 1/24/05 | Vysis, Inc. | UroVysion™ Bladder Cancer Kit |
| P040017 | 3/7/05 | Bayer Healthcare, LLC | ADVIA Centaur® HAV Total Assay |
| P040005 | 5/3/05 | DakoCytomation California | DakoCytomation Her2 FISH pharmDx™ Kit |
| P030049 | 5/26/05 | Bayer Healthcare, LLC | ADVIA Centaur® HBsAg ReadyPack Reagents |
| P040011 | 6/27/05 | DakoCytomation California | DakoCytomation c-Kit pharmDx™ |
| P980022/S011 | 7/18/05 | Medtronic MiniMed | Guardian® RT |
| Document # | Date | Applicant | Device |
|---|---|---|---|
| K041875 | 12/13/04 | Vysis, Inc. | Vysis® Autovysion™ System |
| K042279 | 12/23/04 | Affymetrix, Inc. | Affymetrix GeneChip® Microarray Instrumentation System |
| K042259 | 12/23/04 | Roche Molecular Systems | AmpliChip CYP450 Test |
| K042613 | 4/18/05 | PreAnalytiX GMBH | PAXgene™ Blood RNA System |
| K043011 | 5/9/05 | Tm BioScience Corp. | Tag-It™ Cystic Fibrosis Kit |
| K041847 | 5/19/05 | Wako Chemicals USA, Inc. | Wako LBA AFP-L3 |
| Document # | Date | Applicant | Device |
|---|---|---|---|
| K051713 | 7/6/05 | Idaho Technology, Inc. | JBAIDS Anthrax Detection System |
| K051794 | 8/16/05 | Office of the Surgeon General, U.S Army | Gamma Phage Anthracis Lysis Assay |
| K051824 | 8/18/05 | Third Wave Technologies, Inc. | Invader® UGT1A1 Molecular Assay |
In FY 05, OIVD issued eight guidance documents which are listed below. Among the eight, five are Special Controls guidances. In addition to these IVD-specific guidances, there are other non-IVD-specific guidances that would also apply to IVDs. All of these guidance documents and other previously issued guidance documents are available on the World Wide Web (CDRH homepage: http://www.fda.gov/cdrh) which provides easy access to the latest information, operating policies and procedures. They may also be obtained from the Division of Small Manufacturers International and Consumer Assistance (DSMICA, HFZ-200). To contact DSMICA, call 800-638-2041 or 240-276-3103; Fax 240-276-3151; Email dsmica@cdrh.fda.gov or write to DSMICA (HFZ-200, Food and Drug Administration, 1350 Piccard Drive, Rockville, Maryland 20850-4307).
OIVD staff continue to play a significant role in both domestic and international standards committees. Our staff members are liaisons and chairpersons of standards committees for a wide range of devices for a variety of standards organizations including ISO and CLSI. OIVD is committed to the standards development process, and we believe that the development of scientifically sound standards will allow us to efficiently review premarket applications for both existing and innovative technologies. A complete list of OIVD staff involved in standards activities is given in Appendix B.
| Title | Date Recognized |
|---|---|
| Laboratory Automation: Data Content for Specimen Identification, AUTO7-A | April 2005 |
| Standard Specification for Low-Level Protocol to Transfer Messages Between Clinical Laboratory Instruments and Computer Systems, LISI-A | April 2005 |
| Specification for Transferring Information Between Clinical Laboratory Instruments and Information Systems, 2 nd Edition, LIS2-A2 | April 2005 |
| Standard Guide for Selection of a Clinical Laboratory Information Management System, LIS3-A | April 2005 |
| Standard Guide for Documentation of Clinical Laboratory Computer Systems, LIS4-A | April 2005 |
| Standard Specification for Transferring Clinical Observation Between Independent Computer Systems, LIS5-A | April 2005 |
| Standard Practice for Reporting Reliability of Clinical Laboratory Information Systems, LIS6-A | April 2005 |
| Standard Specification for Use of Bar Codes on Specimen Tubes in the Clinical Laboratory, LIS7-A | April 2005 |
| Standard Guide for Functional Requirements of Clinical Laboratory Information Management Systems, LIS8-A | April 2005 |
| Standard Guide for Coordination of Clinical Laboratory Services within the Electronic Health Record Environment and Networked Architectures, LIS9-A | April 2005 |
Historically, the ODE Annual Report has included premarket combined data for both ODE and OIVD. For FY 05, OIVD and ODE have generated separate annual reports. This is the first report that includes only data for OIVD. For additional information on the premarket review program please refer to the ODE annual report. In this part, first, we present the major submissions1 received in OIVD for FY 04 and FY 05. For these submissions (known as “the receipt cohort”), we provide our review performance for Premarket Approval Applications (PMAs), PMA supplements, Premarket Notifications (510(k)s), Investigational Device Exemptions (IDEs), Pre-IDEs, Humanitarian Device Exemptions (HDEs), and Request for Classification Information (513(g)s). For PMAs and 510(k)s, in addition to review performance data, we also provide our progress toward meeting MDUFMA performance goals. In the second part of this section, we provide information on the number of major submissions processed in FY 05 (known as “the decision cohort”).
As shown in Table 1, during FY 05, OIVD received 666 major submissions, a slight increase from 659 submissions received in FY 04.
Of the 6 original PMAs and 1 panel track supplement received in FY 05, only 1 was granted expedited status. Similarly, in FY 04, only 2 out of 14 original PMAs and panel track supplements submitted to OIVD received expedited status. In FY 05, none of the 6 original PMAs were submitted as modular PMAs as compared to 3 out of 14 (21%) PMAs submitted as modular PMAs in FY 04.
Of the 84 PMA supplements received in OIVD in FY 05, 18 were categorized as 180-day PMA supplements, up from 10 in FY 04. The number of fee paying 180-day supplements, however, remains stable between FY 05 (8) and FY 04 (8).
A total of 11 requests were received and processed as Real-time PMA supplements in FY 05, slightly down from 17 in FY 04. Most applicants chose telephone conferencing versus a face-to-face meeting or a videoconference.
OIVD received 17 Special PMA supplements in FY 05, an increase from 7 in FY 04.
In addition to 180-day, Special and Real-time supplements, OIVD also received 37 30-day notices/135-day supplements in FY05, a slight increase from the 34 30-day notices/135-day supplements received in FY 04.
Of the 520 510(k)s received in FY 05, 455 were submitted as traditional 510(k)s, 16 were submitted as abbreviated 510(k)s, and the remaining 49 were Special 510(k)s. One 510(k) was granted expedited status in FY 05.
Since FY 03, OIVD has seen a significant increase in the number of 513(g)s received every year. A 513(g) is a request for information regarding FDA regulatory requirements applicable to a device. Thirty-one and twenty-six 513(g)s were received in FY 04 and FY 05, respectively. This almost doubled the number of 513(g)s received in the two previous years.
OIVD received a slight increase in the number of original IDEs and IDE supplements between FY 04 and FY 05. In FY 05, OIVD received and processed 6 original IDEs and 23 IDE supplements as compared to 4 original IDEs and 14 IDE supplements in FY 04.
In FY 05, OIVD received 1 original HDE and no HDE supplements as compared to 1 original HDE and 1 HDE supplement in FY04.
| TYPE OF SUBMISSION | 2004 OIVD |
2005 OIVD |
|---|---|---|
| Original PMAs | 14 | 6 |
| PMA Supplements | 69 | 84 |
| Original IDEs | 4 | 6 |
| IDE Supplements* | 14 | 23 |
| 510(k)s | 525 | 520 |
| Original HDE | 1 | 1 |
| HDE Supplements | 1 | 0 |
| 513(g)s | 31 | 26 |
| Total | 659 | 666 |
* Pre-IDEs reported separately
Figures 1 and 2 below provide the OIVD review performance for PMAs filed in FY 01 to FY 04. The data for FY 05 was not included because a significant number of PMA submissions received in FY 05 are still under review and a final decision has not been issued. Therefore, the data for FY 05 will be presented in the next OIVD annual report. Additional PMA related performance is presented in Figures 3-5.
Figure 1: Average Total FDA Review Days from Filing to Approval (excluding withdrawals) For All Original and Panel Track PMA Supplements
Average Total FDA Review Time For All Original and Panel-Track Supplements
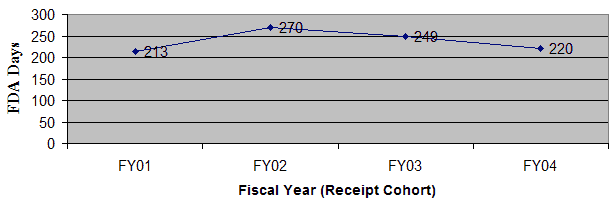
Figure 2: Average Total Elapsed Days from Filing to Approval (excluding withdrawals) for All Original and Panel Track PMA Supplements
Average Total Elapsed Time For All Original PMAs and PT Supplements
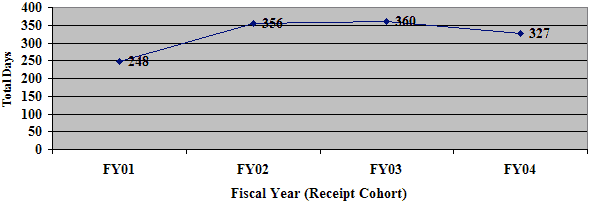
Figure 3: Average Total FDA Days from Receipt to Final Decision for all 180-day PMA Supplements
Average Total FDA Review Time to Final Decision for all 180-Day PMA Supplements
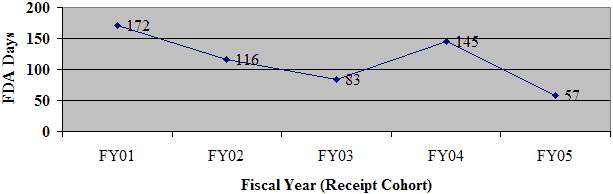
Figure 4: Average Total Elapsed Days from Receipt to Final Decision for All 180-day PMA supplements.
Average Total Elapsed Time to Final Decisions for all 180-Day PMA Supplements
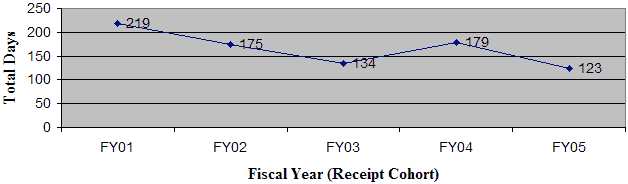
No original PDPs or PDP supplements were approved in FY 05. Note that a PDP that has been “declared complete” is considered to have an approved PMA.
As shown in Figure 6, the average FDA review time from receipt to final decision has declined slightly from FY 01 through FY 05. For FY 05, the average OIVD review time was 61 days, up from 59 days in FY 04. Similarly, the average total elapsed time increased from 84 days in FY 04 to 88 days in FY 05 (Figure 7).
Figure 6: Average Total FDA Time from Receipt to Final Decision
OIVD 510(k)s: Average FDA Time to Final Decision - As of December 31, 2005
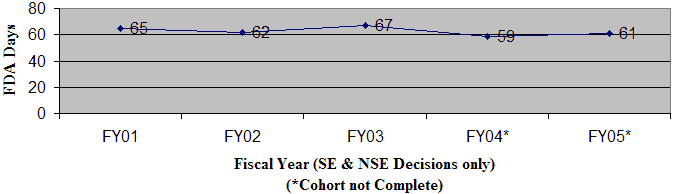
Figure 7: Average Total Elapsed Time from Receipt to Final Decision
OIVD 510(k)s: Average Total Time to Final Decision - As of December 31, 2005
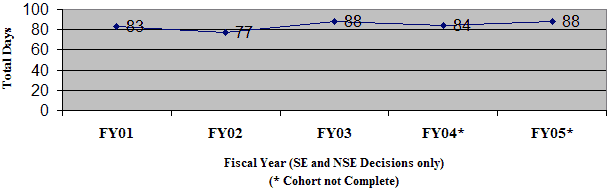
During FY 05, OIVD received three 510(k)s reviewed by third-party organizations under the Accredited Persons provisions (Section 523) of the Federal Food, Drug, and Cosmetic Act.
The average days from the time FDA received the completed 510(k) from the third-party reviewer to the time FDA issued the final decision to the 510(k) holder was 40 days in FY 05, unchanged from FY 04 (Figure 8).
CDRH continued to take steps during FY 05 to improve the quality and consistency of third-party reviews and facilitate timely CDRH action on these submissions. CDRH conducted a training session for ODE/OIVD staff on October 22, 2004 in Rockville, Maryland, and for third-party reviewers on October 26-27, 2004 in Gaithersburg, Maryland. CDRH also conducted telephone conferences with all third-party organizations in January and April 2005 to provide a routine forum for discussing issues and answering questions.
Information on the 510(k) Accredited Persons Program is available on the Center’s third-party review web page at http://www.fda.gov/cdrh/thirdparty/.
Figure 8: 510(k)s Received By OIVD with a Third-Party Review
510(k)s Received by OIVD With a Third-Party Review - As of 31-Dec-2005
OIVD received 1 original HDE in FY 05 and it is still under review.
In FY 05, OIVD received 6 original IDEs and 6 decisions were made. One hundred percent of all original IDE decisions were issued within 30 days in FY 05. The average review time was 16 days (Figure 9).
Figure 9: Average FDA Review Time for Original IDEs
Average FDA Review Time for All Original IDEs
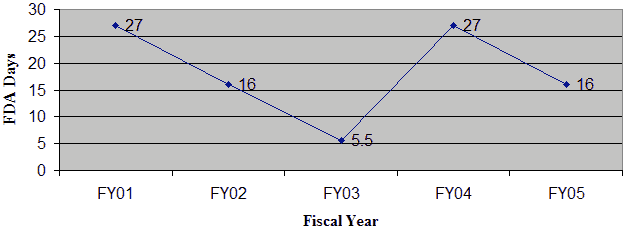
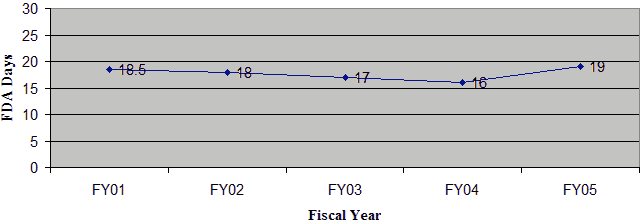
In FY 05, 100% of the IDE supplements received were reviewed within the 30-day statutory timeframe. As shown in Figure 10, the average review time for IDE supplements has remained fairly constant.
Figure 10: Average FDA Review Time for All IDE Supplements
Average FDA Review Time for All IDE Supplements
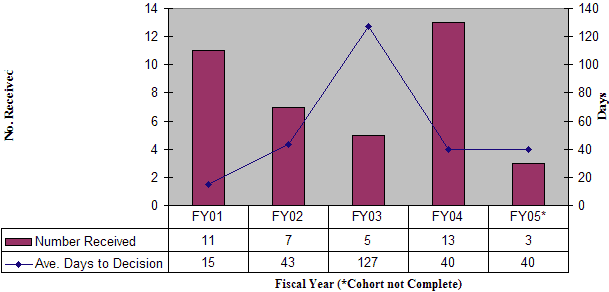
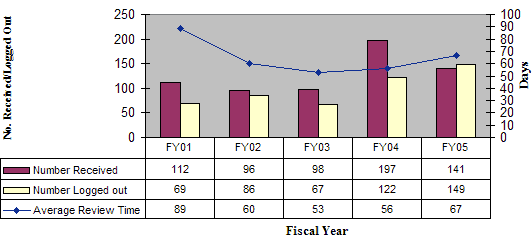
During FY 05, OIVD reviewed 141 pre-IDEs. Based on these reviews, guidance for the pre-IDE submissions were provided to the sponsors through meetings with the sponsors, letters, fax, or by phone. The number of pre-IDE submissions has increased steadily every year due primarily to increasing awareness of the existence and usefulness of the program, as well as increasingly complex devices and combination products. Review times for pre-IDEs have actually decreased over the period of FY 01 through FY 05 despite significantly increased demands on the program (Figure 11).
Figure 11: Pre-IDE Submissions and Average Review Days
Pre-IDE Submissions Received / Logged Out by OIVD
FDA provides regular updates on MDUFMA performance and these reports are available at the following website: www.fda.gov/cdrh/mdufma. Overall, OIVD has made excellent progress in implementing MDUFMA and is achieving nearly all of the performance goals. CDRH has worked hard to communicate the new requirements and challenges of MDUFMA to its staff and stakeholders. To ensure that the implementation of the new law proceeds smoothly, CDRH has worked with its stakeholders and is confident that the implementation of MDUFMA will result in significant benefits to industry, health care professionals, and, most importantly, patients.
The table below summarizes the actions that OIVD completed in fiscal years 2004-2005 (i.e., the “decision cohort”). Note that decisions may be made in one fiscal year for an application that was submitted in a previous fiscal year.
Table 2: Major Submissions Completed FY 04 - FY 05
| TYPE OF SUBMISSION | 2004 OIVD |
2005 OIVD |
|---|---|---|
| Original PMAs | 13 | 11 |
| PMA Supplements | 41 | 28 |
| Original IDEs | 4 | 6 |
| IDE Amendments | 0 | 0 |
| IDE Supplements | 12 | 23 |
| 510(k)s | 541 | 504 |
| Original HDE | 1 | 0 |
| HDE Supplements | 1 | 0 |
| Total | 613 | 572 |
In FY 05, OIVD completed 20 PMA actions on 11 original PMA submissions. These actions included 5 filing decisions, 4 major deficiency decisions, and 11 approval/approvable/not approvable decisions.
Of the 11 approval/approvable/not approvable decisions made in FY 05 on original PMAs, 9 were approval orders, 1 was approvable and 2 were not approvable. Of the 9 approvals, none were for expedited PMAs. See Part 1 (ADVANCES IN PATIENT CARE) for a complete list of PMA approvals.
In FY 05, OIVD completed 49 PMA supplement actions on 28 PMA supplement submissions. These actions included 2 panel-track PMA supplement filing decisions, 6 not approvable decisions, 2 major deficiency decisions, 11 approvable decisions and 28 approval decisions in FY 05.
OIVD completed 504 510(k) actions in FY 05. These actions included 434 substantially equivalent decisions, 16 not substantially equivalent decisions, and 54 other decisions such as withdrawn or deleted.
During the fiscal year, 53 Special 510(k)s received final decisions (49 were found substantially equivalent, 0 were found not substantially equivalent, and the remaining 4 had other decisions).
Sixteen abbreviated 510(k)s received final decisions (15 substantially equivalent, 0 not substantially equivalent, and 1 other decision).
OIVD made final decisions on 3 “third party” 510(k)s in FY 05, 3 substantially equivalent.
The percentage of IDEs approved on the first review cycle was 100% in FY 05. Like original IDEs, the percentage of IDE supplements reviewed within the 30-day statutory timeframe was 100% in FY 05.
The Food and Drug Administration Modernization Act of 1997 (FDAMA) amended Section 513(f) (21 U.S.C. 360c(f)) to provide a new mechanism to reclassify statutorily classified class III products. This provision, which is referred to as the Evaluation of Automatic Class III Designation provision (also known as "de novo" or "risk-based" classification), is intended to apply to low risk products that have been classified as class III because they were found not substantially equivalent (NSE) to any identifiable predicate device. The process permits the Secretary (FDA, by delegation) to reclassify certain low risk devices into class I or II on the basis of established risk-based classification criteria. See Part 1 (ADVANCES IN PATIENT CARE) for a complete list of Automatic Evaluation of Class III Designated products.
The integration of post-market monitoring of product quality and safety in OIVD is continuing to evolve to better protect public health. OIVD continues to apply a risk-based approach for management of inspection and enforcement actions so that our efforts will have the maximum effect on the public health. OIVD is responsible for evaluating and monitoring compliance and surveillance programs covering regulated industry; evaluating industry quality systems, quality control, and testing programs to assure compliance with regulations; and reviewing and monitoring advertising and promotion of IVDs to determine compliance with regulations. We interact with Agency field offices and other Agency staff relating to legal actions, case development, and product recalls. In FY 2005, there were 7 Class I Recalls, 3 Warning Letters and no Seizures. A brief description of each action is listed below. In addition, there were 9,605 MDR Reports for FY 2005.
Class 1 Recalls on the web: http://www.fda.gov/ora/compliance_ref/recalls/recallpg.html
| Applicant | Device Name | Date Initiated / Link |
|---|---|---|
| Ortho-Clinical Diagnostics | VITROS® Immunodiagnostic HBsAg Confirmatory Kit | 12/15/05 |
| bioMérieux, Inc. | VeriCal® Calibrator Sets | 8/19/05 |
| Abbott | Blood Glucose Meters | 6/7/05 |
| LifeScan, Inc. | Blood Glucose Meters | 4/11/05 |
| bioMérieux, Inc. | Simplastin® HTF Tissue Reagent | 3/7/05 |
| MicroScan | MicroScan® Rapid Pos Inoculum Broth | 2/7/05 |
| Becton Dickinson | ProbeTec™ ET Urine Processing Kit | 1/10/05 |
Warnings Letters on the web: http://www.fda.gov/foi/warning.htm
| Applicant | Date Issued | Subject |
|---|---|---|
| BioImagene, Inc. | 5/25/05 | Lacks Approved Premarket Application/Adulterated |
| Access Genetics | 8/1/05 | Lacks Premarket Notification Clearance/Adulterated/Misbranded |
| Tepnel Diagnostics, Ltd. | 8/26/05 | Lacks Premarket Approval Application/ Adulterated |
Under the Application Integrity Program (AIP), OIVD has considered cases concerning the integrity of data submitted to the agency in premarket applications. During FY 05, we placed one application on Integrity Hold and removed the Integrity Hold on another application.
Congress passed the Clinical Laboratory Improvement Amendments (CLIA) in 1988, establishing quality standards for all laboratory testing to ensure the accuracy, reliability and timeliness of patient test results regardless of where the test was performed. The categorization of commercially marketed in vitro diagnostic tests under CLIA has been the responsibility of the FDA since January 31, 2000. FDA performs the CLIA complexity categorization that includes the assignment of these test systems to one of three CLIA regulatory categories (waived, moderate and high) based upon their level of complexity. A CLIA waiver determination may be granted to: 1) the nine generic tests waived by the regulation, 42 CFR 493.15(c); 2) any test system for which the manufacturer or producer applies for waiver if that test meets the statutory criteria and the manufacturer provides scientifically valid data verifying that the waiver criteria have been met; and 3) test systems cleared by the FDA for home use.
During FY 05, OIVD performed categorizations on a total of 1962 tests including 429 waived, 1288 moderate, and 245 high tests. The number of waived tests increased more than two-fold from FY 04. CLIA categorizations for previous fiscal years are shown in the graph below, Figure 12.
Figure 12. CLIA Categorizations
CLIA Categorizations
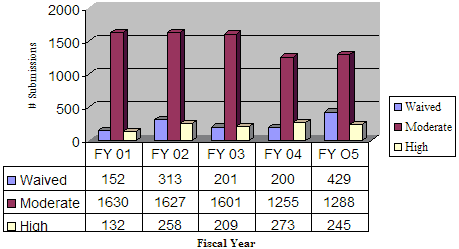
An on-line, searchable CLIA database is available that contains the commercially marketed in vitro test systems categorized by the FDA since January 31, 2000, and tests categorized by the Centers for Disease Control and Prevention (CDC) prior to that date. More information on the CLIA program can be found at http://www.fda.gov/cdrh/clia/index.html.
OIVD continues to be involved in several resource-intense initiatives related to national bioterrorism preparedness and response. OIVD established liaisons to collaborate with other government agencies and the military to prepare for and assume regulatory responsibilities applicable to medical devices that are critical to bioterrorism preparedness efforts. ODE/OIVD is currently developing guidance and procedures for timely premarket review and approval of these devices.
OIVD cleared two devices for the detection of Bacillus anthracis: the Joint Biological Agent Identification and Diagnostic System (JBAIDS) Anthrax Detection System (an anthrax immunoassay) sponsored by Idaho Technology and a Gamma Phage Lysis assay specific for Bacillus anthracis manufactured by the Army Medical Research Institute of Infectious Diseases (USAMRIID) and distributed by CDC. OIVD participated in inter-agency meetings related to "Evaluation of Pre-Screening Technologies" for possible biological agents/threats, on the TOPOFF 3 exercise, and in BioShield initiatives such as the Emergency Use Authorization Working Group lead by HHS and the development of the Emergency Use Authorization of Medical Products Draft Guidance.
OIVD’s bioterrorism preparedness activities also formed the building blocks for the office in helping the government prepare for pandemic flu and other potential health emergencies as this becomes an area of increasing importance.
In an effort to standardize the 510(k) submission, reduce premarket review effort and develop a better premarket/post market balance more in line with the TPLC regulatory concept, OIVD continued developing the Turbo 510(k) eSubmission program. An eSubmission guides industry to submit only scientifically and administratively complete applications, making it easier to meet MDUFMA goals. In addition, this program preserves institutional memory, enhances knowledge management and increases consistency of decision-making regarding product clearances.
Turbo 510(k) was piloted in FY 04 and significant gains were made in the enhancement of the program through direct feedback from the industry volunteers who submitted their 510(k) using the eSubmission.
In an effort to provide stakeholders with the scientific / regulatory basis for FDA’s decisions and to be consistent with CDRH’s center-wide “Knowledge Management” and “Transparency” initiatives, OIVD developed and implemented the use of a standardized Premarket Decision Summary Template across all the OIVD divisions.
The Premarket Decision Summary Template summarizes the basis on which an in vitro diagnostic device was cleared under a 510(k) submission. OIVD implemented the use of this standardized premarket Decision Summary Template in August 1, 2003. The decision summaries have been continuously posted on the OIVD webpage and the public has full access to them through the 510(k) database. CDRH has been receiving positive feedback from regulated industry, the professional community, and consumers because the decision summaries have given them the ability to make informed decisions regarding the use of these in vitro diagnostic devices.
In FY 05, an Immunology Devices panel meeting was held. The panel reviewed the PMA, made recommendations, and voted on an application for a laboratory assay designed to measure levels of neural thread protein in urine specimens from patients presenting with cognitive complaints or other signs and symptoms of suspected Alzheimer’s disease. The panel unanimously recommended that the PMA be found not approvable. Refer to the ODE annual Report for more information on advisory panels.
OIVD is responsible for the program areas where in vitro diagnostic devices are regulated from conception to obsolescence. This Appendix provides summary information on major program areas administered by parallel offices (ODE, OC, and OSB). OIVD is responsible for carrying out its activities in a consistent manner with all of these programs area. Please see below for a summary of the OC and OSB program areas; and refer to the ODE Annual Report, Appendix A, for a brief description of the premarket programs.
The Enforcement / Compliance program stimulates awareness within the agency of the need for prompt and positive action to ensure compliance by regulated industries; works to ensure an effective and uniform balance between regulatory compliance and agency responsiveness to consumer needs; acts as liaison with other federal agencies on agency compliance matters, and encourages an effective and appropriate balance between voluntary and regulatory compliance; evaluates and coordinates proposed legal actions to ascertain compliance with regulatory policy and enforcement objectives; evaluates and coordinates proposed legal actions to ascertain compliance with regulatory policy and enforcement objectives.
The Food and Drug Administration Modernization Act of 1997 (FDAMA) modified Postmarket Surveillance (PS) requirements under section 522 of the Act. Postmarket surveillance means the active, systematic, scientifically valid collection, analysis, and interpretation of data or other information about a marketed device. The data can reveal unforeseen adverse events, the actual rate of anticipated adverse events, or other information necessary to protect the public health.
Postmarket issues may be identified through a variety of sources, including analysis of adverse event reports, a recall or corrective action, warning letter, a seizure, reports from other governmental authorities, or the scientific literature. The guidance document “Postmarket Surveillance Under Section 522 of the Federal Food, Drug and Cosmetic Act” can be viewed at: http://www.fda.gov/cdrh/osb/guidance/316.html
Medical Device Reporting (MDR) is the mechanism for the Food and Drug Administration to receive significant medical device adverse events from manufacturers, importers and user facilities, so they can be detected and corrected quickly. User Facilities (e.g., hospitals, nursing homes) are required to report suspected medical device related deaths to both the FDA and the manufacturers. User facilities report medical device related serious injuries only to the manufacturer. If the medical device manufacturer is unknown, the serious injury is reported by the facility to FDA. On July 31, 1996, the new Medical Device Reporting (MDR) regulation became effective for user facilities and device manufacturers.
The Medical Product Safety Network (MedSun) is a pilot program launched in 2002 by CDRH. The primary goals for MedSun are to identify, understand and share information about problems with the use of medical devices. MedSun plays an important role in FDA's post-market surveillance effort. Hospitals, nursing homes, and other healthcare facilities are currently required to report medical device problems under the Safe Medical Devices Act (SMDA). MedSun provides a secure, Internet-based data entry system that automates this process and helps gather other additional data that can help FDA, device manufacturers, and clinical facilities proactively address safety concerns before serious injuries or deaths occur.
Bast RC, Lilja H, Urban N, Rimm DL, Fritsche H, Gray J, Veltri R, Klee G, Allen A, Kim N, Gutman S, Rubin MA, Hruszkewycz A. Translation Crossroads For Biomarkers. Clin Cancer Res 2005; 11: 6103-6108.
Harper CC, Philip R, Robinowitz M, and Gutman SI. FDA Perspectives On Pharmacogenetic Testing. Expert Review of Molecular Diagnostics 2005; 5: 643-648.
Hackett JL and Gutman SI. Introduction to the Food and Drug Administration
(FDA) Regulatory Process. Journal of Proteome Research 2005; 4:1110-1113.
Hausman ED and Altaie SS. Regulatory Aspects of Total Product Life Cycle. Diabetes Technology & Therapeutics 2004; 6:761-766.
Shi L, Tong W, Fang H, Scherf U, Han J, Puri RK, Frueh FW, Goodsaid FM, Guo L, Su Z, Han T, Fuscoe JC, Xu, ZA, Patterson TA, Hong H, Xie Q, Perkins RG, Chen JJ, Casciano DA. Cross-Platform Comparability of Microarray Technology: Intra-Platform Consistency and Appropriate Data Analysis Procedures Are Essential, BMC Bioinformatics 2005 Jul 15:6 Suppl 2:S12.
Altaie SS. Pre-IDE process in IVDs. Annual Association of Medical Diagnostic Manufactures and FDA 510(k) workshop, Rockville, MD, April 19-20, 2005.
Altaie SS. OIVD’s Turbo 510(k) update, 32nd Annual Meeting of Association of Medical Diagnostics Manufacturers, Rockville, MD, April 21-22, 2005.
Altaie SS. 510(k) Decision Summaries and eSubmissions. Annual Association of Medical Diagnostic Manufactures and FDA 510(k) workshop, Rockville, MD, April 19-20, 2005.
Altaie SS. Critical Path Initiative in Medical Devices. RAPS 2005 Annual Conference and Exhibition, Baltimore, MD, October 16-19, 2005.
Akolkar PN and Hojvat S. Migration Studies. 4th Annual CBER/CDRH Harmonized Best Practices Workshop for In Vitro Diagnostics, Rockville, MD, June 30, 2005.
Becker RL. Biomarkers and Surrogate Endpoints: The FDA Perspective. IBC Biomarker Pipeline Conference, Boston, MA, March 14-15, 2005.
Becker RL. Urinary Neural Thread Protein Test For Alzheimer's Disease. FDA Staff Roundup on Alzheimer's Disease: Diagnosis to Treatment, Rockville, MD, August 31, 2005.
Benson CC. CLIA Update. RAPS Meeting, Washington, DC, October 12, 2004.
Chan MM. FDA Regulation of Biochips. 2nd KFDA-KRIBB Joint International Symposium on “International Harmonization on Biopharmaceuticals – Recent Trends in Research, Development and Evaluation Technology,” Seoul, South Korea, October 26-30, 2004.
Chan MM. Regulatory Perspectives in Pharmacogenomics. 1st Spanish Congress on Pharmacogenetics and Pharmacogenomics, Valencia, Spain, January 22-26, 2005.
Chan MM. Regulation of Pharmacogenomic Devices – OIVD Perspectives. University of Tokyo sponsored International Symposium on “Frontiers in Drug Development,” Tokyo, Japan, February 16-19, 2005.
Gutierrez A. FDA Regulation Opportunity or Hindrance, Alcohol Biosensors Annual Progress Meeting, Rockville, MD, July 21, 2005.
Gutierrez A. 510(k) Review II, DSMICA Workshop, Los Angeles, CA, September 30, 2005.
Gutman SI. Panel Member – Regulating and Reimbursing New Genetic Technologies: “What Manufacturers and Clinical Laboratories Need to Know,” American Association of Clinical Chemistry Audio conference, Atlanta, GA, October 13, 2004.
Gutman SI. Keynote Address: An Update on FDA Regulation of Medical Devices – Applications to the Management of Diabetes. Diabetes Technology Meeting, Philadelphia, PA, October 29, 2004.
Gutman SI. Review of Regulatory Framework for Chem/Bio Defense Diagnostic Countermeasures. Chem/Bio Warfare Defense Panel of the Threat Reduction Advisory Committee to the Department of Defense, Arlington, VA, November 5, 2004.
Gutman SI. FDA Perspective on Diagnostics – How to Prepare a Premarket Submission. Molecular Diagnostics – Market, Regulatory and Payer Requirements for Commercial Success, CBI and Boston Health Care Associates, Washington, DC, November 19, 2004.
Gutman SI. Approval of Pharmacogenomics Tests. Pharmacogenetics Research Network (PGRN) Steering Committee, Orlando, FL, March 2, 2005.
Gutman SI. FDA Regulation: Opportunity or Obstacle. Biomedical Marketing Association Gutman SI. Moderator: Experience with VGDS and Required PGx Submissions. Breakout Session, Pharmacogenomics in Drug Development and Regulatory Decision Making, Drug Information Association, Bethesda, MD, April 12, 2005.
Gutman SI. Update on OIVD. Annual Meeting of the Association of Medical Diagnostics Manufacturers, Rockville, MD, April 21, 2005.
Gutman SI. Genomics – Translating the Science into Regulatory Decision-Making – Interactive Round Table Discussion. FDA Science Forum, Washington, DC, April 27, 2005.
Gutman SI. The New CLIA waiver criteria. AACC, ACLA and ASCLS Audio Conference, Rockville, MD, May 4, 2005.
Gutman SI. Developing Standards for the Exchange of Genomic and Proteomic Data from Clinical Trials – The Regulatory Game Plan. American Society of Clinical Oncology, Annual 2005 Meeting, Orlando, FL, May 16, 2005.
Gutman SI. Panelist: ThinkTank II: Pharma/FDA Dialogue. The Biomarker World Congress 2005, Philadelphia, PA, May 24, 2005.
Gutman SI. Regulatory Issues in the Co-Development of Drugs and Diagnostics – Don’t Forget the Diagnostic! The Biomarker World Congress 2005, Philadelphia, PA, May 25, 2005.
Gutman SI. Panelist: ThinkTank IV: Exploiting the Synergy between Pharma and Diagnostic Biomarker Research. The Biomarker World Congress 2005, Philadelphia, PA, May 25, 2005.
Gutman SI. FDA Regulatory Perspectives – In Vitro Diagnostic Regulation. NCI Biosensor Symposium, Bethesda, MD, June 8, 2005.
Gutman SI. IVD Update. Reality of Regulatory Affairs, 8th Annual FDA-OCRA Educational Conference, Irvine, CA, June 16, 2005.
Gutman SI. FDA Regulation of In Vitro Diagnostic Devices: How To Turn The Critical Path Into A Yellow Brick Road. Plenary Session on Biomarkers, 13th Specialized Programs of Research Excellence (SPORE) Investigator’s Workshop, Washington, DC, July 9, 2005.
Gutman SI. Moderator: Clinical Validation. Microarrays in Transcriptional Profiling: A Joint John Hopkins/FDA/PhRMA Workshop, Rockville, MD, July 21, 2005.
Gutman SI. IVD Regulation by the FDA in the United States. U.S Regulation and Reimbursement of In Vitro Diagnostic Products, IFCC/ACCC Annual Meeting, Orlando, FL, July 24, 2005.
Gutman SI. Regulatory Issues for Genomic Tests: Shortening the Critical Path to market. Global Genomic Personalized Medicine – EduTrak, IFCC/AACC Annual Meeting, Orlando, F, July 26, 2005.
Gutman SI. The New Waiver Criteria: What They Mean to the Clinical Laboratory Industry Healthcare Forum, IFCC/AACC Annual Meeting, Orlando, FL, July 27, 2005.
Gutman, S.I., From Clinical Trials to Drug Approval. Genetic Alliance 2500 Annual Conference, Rockville, MD, July 29, 2005.
Gutman SI. Progress and Pitfalls in Development and Validation of Biomarkers – Regulatory Aspects. Standards, Methods, Assays, Reagents and Technologies (SMART) for Early Cancer Detection and Diagnostics, National Institute of Standards and Technology, Gaithersburg, MD, August 18, 2005.
Gutman SI. Panelist, Symposium I : Emerging Pharmacogenomic Tools and Their Utility in Benefit/Risk Assessment in Clinical Trials. 34th Annual Meeting American College of Clinical Pharmacology, Rockville, MD, September 11, 2005.
Gutman SI. and Hojvat S. FDA update and ASR’s/New Technology Submissions. The 8th Annual FDA-OCRA Educational Conference, Irvine, CA, June 16, 2005.
Hackett JL. FDA Perspective: Clinical, Regulatory, and QA Issues Related to Genomics. Regulatory Affairs Professional Society 2004 Annual Conference. Washington, DC, October 12, 2004.
Hackett JL. Pharmacogenomics: FDA and In Vitro Diagnostics. Personalized Medicine. Accelerating Dx/Rx Product Development, Center for Business Intelligence, Philadelphia, PA, June 27, 2005.
Harper CC. FDA Perspectives on Pharmacogenetic Testing. CHI 7th Annual Systems Biology Conference, San Francisco, CA, June 15, 2005.
Harper CC. The Roche AmpliChip CYP450 Microarray: A Summary of the Regulatory Process, Senate HELP Committee Briefing, Washington, DC, January 26, 2005.
Hojvat S and Toth-Allen J. In Vitro Diagnostic (IVD) Devices: How They Differ from Other Devices & How FDA Regulates Them. Association of Clinical Research Professional (ACRP), Orlando, FL, April 5, 2005.
Hojvat S. Human Specimen Repositories Requirements of 21 CFR Parts 50 & 56: Biological Specimens and Personal Data. Critical Path to New Medical Products: The Challenges In Protecting Human Subjects, Houston, TX, April 22, 2005.
Hojvat S. Office of In Vitro Diagnostic Device Evaluation and Safety General Compliance Update. The 8th Annual FDA-OCRA Educational Conference, Irvine, CA, June 16, 2005.
Hojvat S. FDA’s Current Thinking on GLP/GCP: Building Quality Into Device Clinical Trials. Quality Device Trials, “Microarrays in Transpotional Profiling”, DDA/John Hopkins/PhRMA, Rockville, MD, July 21, 2005.
Hojvat S. 510 (k) Review (1), DSMICA Workshop, Los Angeles, CA, September 30, 2005.
Hojvat S. FDA Regulation – Opportunity or Barrier? “Chips to Hits,” Boston, MA,
September 10, 2005.
Hojvat S. Microarrays Used for Pathogen Detection – An FDA Perspective. Boston, MA, September, 2005.
Pastel MS. FDA/CDRH Perspective on Regulatory Requirements for Array Bioinformatics. AACC's Lab 2007: Your Eye to the Future, October 21-22, 2004, Chicago IL.
Poole FM. Bacterial ID Systems: Quality Control and Regulations, an FDA Perspective. Clinical Laboratory Improvement Advisory Committee Meeting, CDC, Atlanta, GA, Feb 15-16, 2005.
Poole FM. Regulatory Framework for New Technologies in Vitro Diagnostic Devices. Biotechnology Seminar, US Military Academy, West Point, NY, May 5-6, 2005.
Scherf U. Pharmacogenomics in Drug Development and Regulatory Decision Making. Drug Information Association, Pharma Meeting, North Bethesda, MD, April 11-13, 2005.
Shively RG. Regulating IVDs for Emerging Infectious Diseases. Regulatory Affairs Professionals Society (RAPS) Annual meeting, Baltimore, MD, October 12, 2004.
Shively RG. Tools to Navigate In Vitro Diagnostic Critical Paths. American Society for Microbiology Biodefense Meeting, Baltimore, MD, March 2005.
Shively RG. Multiplexing with FDA. National Institute for Allergy and Infectious Diseases, NIH Multiplexing workshop, Bethesda, MD, June 27, 2005.
Shively RG. Postmarket Controls. CDC TB Expert Meeting/Workshop, Atlanta, GA, June 27, 2005.
Shively RG. FDA/OIVD Premarket Reviews. CDC (NCID), Atlanta, GA, September 8, 2005.
Simon KA. FDA Perspectives on Pharmacogenetic Testing. IBC's 4th Annual Conference, Molecular Diagnostics and Personalized Medicine, Boston, MA, September 12-15, 2005.
St.Pierre DJ. Introduction to OIVD. ORCA Meeting, Seattle, WA, March 15, 2005.
St.Pierre DJ. Compliance and Postmarketing Trends. Association of Medical Diagnostic Manufacturers, Rockville, MD, April 22, 2005.
St.Pierre DJ. Turbo 510(k) for In Vitro Diagnostic Devices. AdvaMed 15th Annual Submissions Workshop, Arlington, VA, May 24-25, 2005.
St.Pierre DJ. Update on OIVD. Orange County Regulatory Affairs Annual Meeting, Irvine, CA, June 1-4, 2005.
St.Pierre DJ. DSMICA Workshop, Introduction and Update on OIVD “Pre-IDE and De novo Applications,” Los Angeles, CA, September 30, 2005.
| Altaie, Sousan Bautista, Josephine Bernhardt, Patricia Brill, Marieann Brindza, Larry Callaghan, James Carrington, Leonthena Chace, Nina Chappie, Marie Faison, Tremel |
Gutman, Steven Hackett, Joseph Hausman, Ethan Ingram, Kenneth Mcgruder, Louise Moyer, Vicki Pinkos, Arleen Poole, Freddie Reeves, James Rheinheimer, Douglas |
Robinowitz, Max Scherf, Uwe Selepak, Sally Shively, Roxanne Tezak, Zivana Valentine, Fredricka Wright, Kathleen Wilbon, Tonya Wood, Douglas |
OIVD Home Page -- http://www.fda.gov/cdrh/oivd
CDRH’s Home Page -- http://www.fda.gov/cdrh/index.html
Division of Small Manufacturers, International and Consumer Assistance -- http://www.fda.gov/cdrh/consumer/index.html
Federal Advisory Committee Act Database -- http://www.facadatabase.gov/public.asp
FDA’s Home Page -- http://www.fda.gov
Guidance Documents -- http://www.fda.gov/cdrh/guidance.html
Instructions for Submitting Electronic Submissions -- http://www.fda.gov/cdrh/elecsub.html
Least Burdensome Provisions - Activities Related to Implementation -- http://www.fda.gov/cdrh/modact/leastburdensome.html
MDUFMA Home Page -- www.fda.gov/cdrh/mdufma
Panel Meeting Schedules and Summaries -- http://www.fda.gov/cdrh/panel/index.html
Previously Approved/Cleared Device Databases -- http://www.fda.gov/cdrh/consumer/mda/index.html
Recent Device Approvals -- http://www.fda.gov/cdrh/consumer/mda/index.html
Recruitment Brochure for Members and Consultants to the Medical Devices Advisory Committee --http://www.fda.gov/cdrh/ode/advbrochure01.html
Standards of Ethical Conduct -- http://www.usoge.gov/pages/forms_pubs_otherdocs/fpo_files/reference/rfsoc_99.pdf
Third Party Review -- http://www.fda.gov/cdrh/thirdparty
(* - Appointed in FY06)
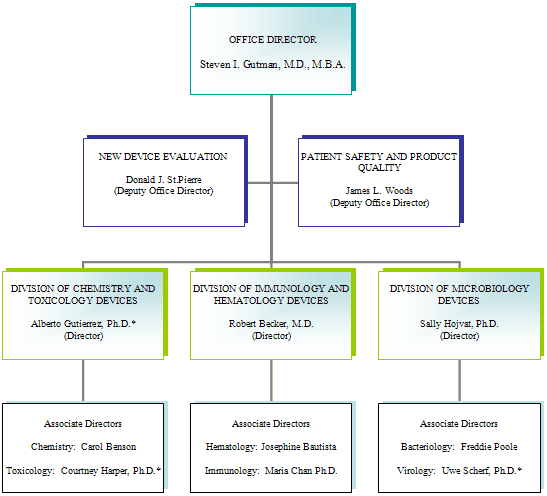
(Reflects staff members on board on the last day of FY 05)
Gutman, Steven
St.Pierre, Don
Woods, James
Altaie, Sousan
Beverly, Patricia
Ellis, Claudette
Fish, Robert
Gavin, Terri
Gonzalez-Licea, Gus
Hackett, Joseph
Hanna, Nancy
Hoard, Renita
Keenan, Rebecca
Latish, Andrea (shared w/OC)
Malcomson, Mercedes
McFarland, Scott
Pastel, Mary (shared w/OSEL)
Schief, Lawanda
Tolbert, Judy
Vashio, Valerie
Warren, Duffy
Wei, Tena
Wilbon, Tonya
Gutierrez, Alberto
Benson, Carol
Bernhardt, Patricia
Callaghan, James
Calvin, Veronica
Caviness, Susanne
Chappie, Ann
Chesler, Ruth
Danishefsky, Avis
Hall, Christina
Harper, Courtney
Hausman, Ethan
Ingram, Kenneth
Millet, Ian
Moyer, Vickie
Pinkos, Arleen
Phillips, Tracy
Reilly, Eugene
Rheinheimer, Douglas
Ritchwood, Jasmine
Shoaibi, Azadeh (shared w/OSB)
Stafford, Elizabeth
Tsai, Miin Rong
Wood, Doug
Hojvat, Sally
Poole, Freddie
Brill, Marieann
Brock, Nadine
Francis, Jacqueline
Gaffey, Claudia
Goldman, Tara
Heyliger, Marian
Rao, Prasad
Scherf, Uwe
Selepak, Sally
Shaikh, Farzana
Shively, Roxanne
Simms, Tom
Simon, Kathleen
Summers, Peter
Tezak, Zivana
Valentine, Fredericka
Whitaker, Kathleen
Wright, Kathy
Becker, Robert
Bautista, Josephine
Chan, Maria
Brindza, Larry
Carlos, Rufina
Carrington, Leonthena
Chace, Nina
Dada, Valerie
Datiles, Therese
Faison, Tremel
Jones, Cecily
Kalush, Francis
Li, Dai
Magruder, Louise
McClain-Bennett, Joan
Menon, Radha
Moore, Deborah
Philip, Reena
Reeves, James
Robinowitz, Max
Stewart, Paula
Weeks, Susan
1 A major submission is defined as an original statutory premarket application that requires FDA’s scientific review and decision.
Updated November 28, 2006
![]()
CDRH Home Page | CDRH A-Z Index | Contact CDRH | Accessibility | Disclaimer
FDA Home Page | Search FDA Site | FDA A-Z Index | Contact FDA | HHS Home Page
Center for Devices and Radiological Health / CDRH




