


FDA
Home Page | CDRH Home Page | Search
| CDRH
A-Z Index | Contact CDRH
![]()
|
| FDA
Home Page | CDRH Home Page | Search
| CDRH
A-Z Index | Contact CDRH
|
|
|
![]()
U.S. Department of Health and Human Services
Public Health Service
Food and Drug Administration
Center for Devices and Radiological Health
![]()
AcknowledgementsThanks to the following organizations for their invaluable assistance in preparing this report:
|
Table of Contents
|
Part 1 – Advances in Patient Care
FETAL OXYGEN MONITOR
MIDDLE EAR SURGICAL IMPLANT
ROBOT-ASSISTED SURGERY
DIGITAL MAMMOGRAPHY
MAPPING THE HEART AND TREATING ARRHYTHMIA
LASER-BASED EYE SURGERY
TREATING GASTROESOPHAGEAL REFLUX DISEASE
FDA Consumer Web Sites
Device Databases
Consumer Information
Original PMA/HDE Approvals for Fiscal Year 2000
Significant Medical Device Breakthroughs
Devices Approved via PMA/HDE
510(k) Clearances or Automatic Evaluations of Class III
Designation Devices (AE)
ODE Guidance Documents
Draft Guidance Documents on the Internet for Comment Purposes
Only
Part 3 – Key Performance Indices
Premarket Approval Applications (PMAs)
Figure 1. Average Review Time for PMA Decision Cohort
Approvals
Figure 2. Original Receipt Cohort PMAs Received and Filed
Figure 3. Receipt Cohort PMA Average Elapsed
Figure 4. Annual Receipts and Actions for PMA Supplement Decision
Cohort
Figure 5. Average Review Time for PMA Supplements
Real-Time Review of PMA Supplements
Product Development Protocols (PDPs)
Modular PMA Review
Humanitarian Device Exemption (HDE) Applications
Investigational Device Exemptions (IDE)
Premarket Notification (510(k)s)
Figure 7. Average 510(k) Review Time for Decision Cohort
Figure 8. Receipts and Actions for 510(k) Receipt Cohorts*
Figure 9. FDA Days from Receipt to Final Action for 510(k)
Receipt Cohorts*
Third-Party Review of 510(k)s
Special 510(k)s
Abbreviated 510(k)s
Significant Medical Device Breakthroughs
Classification Actions
Automatic Evaluation of Class III Designation
Final Reclassification Actions
Class II Exemption Petitions
Final 515(b) Calls for PMAs
Guidance for Industry and Reviewers
Least Burdensome
Significant Jurisdictional Issues Involving Devices in FY 2000
Advisory Panel Activities
ODE Integrity Program
Freedom of Information Requests
Congressional Inquiries
Publications
ODE Vendor Day
Site Visits
In-House Training
Mentoring Program
Other Employee Programs
Minority Recruitment
Computer Tracking Systems
Office Automation
Electronic Submissions
Video Conferencing
World Wide Web Activity
Device Databases
Consumer Information
Table 3. PMA/HDE/IDE/510(k) Submissions Received
Table 4. Original PMA Decision Cohort Performance
Table 5. Original PMA Receipt Cohort Performance
Table 6. PMA Supplement Decision Cohort Performance
Table 7. PMA Supplement Receipt Cohort Performance
Table 8. HDE Submissions Received FY97 – FY00
Table 9. Original HDE Decision Cohort Performance FY97 –
FY00
Table 10. HDE Supplement Decision Cohort Performance FY97 –
FY00
Table 11. Original IDEs FY 96 - FY 00
Table 12. IDE AmendmentsFY 96 - FY 00
Table 13. IDE Supplements FY 96 - FY 00
Table 14. 510(k) Decision Cohort Performance FY 96 - FY 00
Table 15. 510(k) Receipt Cohort Performance FY 96 - FY 00
Appendix A – Summary of Major ODE Programs
Premarket Approval Applications (PMAs)
Product Development Protocols (PDPs)
Humanitarian Device Exemptions (HDEs)
PMA Supplements
Investigational Device Exemptions (IDEs)
IDE Amendments
IDE Supplements
Premarket Notifications (510(k))
Appendix C – Selected FDA Websites


Last year the Office of Device Evaluation (ODE) approved and cleared thousands of devices used to diagnose and treat a wide variety of medical conditions. For a complete listing of newly approved devices, please see Part 2 – INDUSTRY INFORMATION. A new Premarket Approval Application (PMA) approval website describing recently approved devices with patient information is now available at http://www.fda.gov/cdrh/mda/index.html. Below we highlight several medical devices approved during this past fiscal year that we believe will have a major impact on patient care.
 FETAL
OXYGEN MONITOR -- The OxiFirst™ Fetal Oxygen Saturation Monitoring System,
Mallinckrodt, Nellcor Perinatal Business, is a new type of fetal monitor that
measures oxygen saturation in the baby’s blood as a sign of fetal health during labor
and delivery. The OxiFirst™ sensor is inserted into the mother’s uterus and
placed against the temple or cheek of the fetus. The monitor displays fetal oxygen
saturation as percent of oxygen in the fetus’s blood. Oxifirst™ is used along
with conventional electronic fetal monitoring when the fetal heart rate is
"non-reassuring," that is, when the rate indicates that the baby may be in
distress due to lack of adequate oxygen. It is intended for use only on single (not
multiple) fetuses of at least 36 weeks gestation, where the "mother’s
water" has broken and the fetal head is in the normal, head down position for
delivery.
FETAL
OXYGEN MONITOR -- The OxiFirst™ Fetal Oxygen Saturation Monitoring System,
Mallinckrodt, Nellcor Perinatal Business, is a new type of fetal monitor that
measures oxygen saturation in the baby’s blood as a sign of fetal health during labor
and delivery. The OxiFirst™ sensor is inserted into the mother’s uterus and
placed against the temple or cheek of the fetus. The monitor displays fetal oxygen
saturation as percent of oxygen in the fetus’s blood. Oxifirst™ is used along
with conventional electronic fetal monitoring when the fetal heart rate is
"non-reassuring," that is, when the rate indicates that the baby may be in
distress due to lack of adequate oxygen. It is intended for use only on single (not
multiple) fetuses of at least 36 weeks gestation, where the "mother’s
water" has broken and the fetal head is in the normal, head down position for
delivery.
MIDDLE EAR SURGICAL IMPLANT -- The Vibrant Soundbridge, Symphonix
Devices, Inc., is a  surgically implanted hearing device
intended to help adults with moderate to severe nerve hearing loss. The device is
implanted behind the ear in the temporal (skull) bone. It converts sound to mechanical
energy that is transferred to the middle ear. This energy vibrates delicate structures in
the middle ear very much the way normal sound does. The brain interprets the vibrations as
sound. During implant surgery, the surgeon implants a receiver behind the ear. A wire
leads from the receiver to a small electromagnet attached to one of the middle ear bones.
As an alternative to traditional hearing aids, adults with a moderate to severe
sensorineural hearing loss may choose this device. Adults who choose this device should
have already tried using appropriately fitting external hearing aids.
surgically implanted hearing device
intended to help adults with moderate to severe nerve hearing loss. The device is
implanted behind the ear in the temporal (skull) bone. It converts sound to mechanical
energy that is transferred to the middle ear. This energy vibrates delicate structures in
the middle ear very much the way normal sound does. The brain interprets the vibrations as
sound. During implant surgery, the surgeon implants a receiver behind the ear. A wire
leads from the receiver to a small electromagnet attached to one of the middle ear bones.
As an alternative to traditional hearing aids, adults with a moderate to severe
sensorineural hearing loss may choose this device. Adults who choose this device should
have already tried using appropriately fitting external hearing aids.
 ROBOT-ASSISTED
SURGERY -- The Intuitive Surgical da Vinci™ Surgical System, Intuitive
Surgical Inc., is a robotic device that enables a surgeon to perform certain types of
surgery while seated at a console with a computer and video monitor. The surgeon uses
handgrips and foot pedals attached to the computer console to control three robotic arms
that perform the surgery using a variety of surgical tools. The robotic arms, which have a
"wrist" built into the end of the surgical tools, give surgeons additional
manipulation ability during minimal invasive laparoscopic surgery, enabling easier, more
intricate motion and better control of surgical tools. The device is an alternative to
traditional open surgery or minimally invasive manual laparoscopic surgery in an operating
room environment for procedures such as gall bladder disease or gastro-esophageal reflux
disease (severe heartburn).
ROBOT-ASSISTED
SURGERY -- The Intuitive Surgical da Vinci™ Surgical System, Intuitive
Surgical Inc., is a robotic device that enables a surgeon to perform certain types of
surgery while seated at a console with a computer and video monitor. The surgeon uses
handgrips and foot pedals attached to the computer console to control three robotic arms
that perform the surgery using a variety of surgical tools. The robotic arms, which have a
"wrist" built into the end of the surgical tools, give surgeons additional
manipulation ability during minimal invasive laparoscopic surgery, enabling easier, more
intricate motion and better control of surgical tools. The device is an alternative to
traditional open surgery or minimally invasive manual laparoscopic surgery in an operating
room environment for procedures such as gall bladder disease or gastro-esophageal reflux
disease (severe heartburn).
DIGITAL MAMMOGRAPHY -- The
Senographe 2000D Full Field Digital Mammography  System, General
Electric Medical Systems, is an x-ray mammography system that employs a digital
receptor to capture images of the breast. These images can then be printed to film or
displayed on a high-resolution workstation for interpretation by a qualified mammographer.
The device passes x-rays through the breast to a receptor. In this case the receptor
converts the received x-rays into digital signals that can be stored for subsequent
retrieval and display. A mammographer then interprets the displayed image of the breast to
determine whether the breast is normal or whether additional testing is required. This is
an alternative to traditional screening and diagnostic mammography. It can be used
whenever a traditional mammography examination is indicated.
System, General
Electric Medical Systems, is an x-ray mammography system that employs a digital
receptor to capture images of the breast. These images can then be printed to film or
displayed on a high-resolution workstation for interpretation by a qualified mammographer.
The device passes x-rays through the breast to a receptor. In this case the receptor
converts the received x-rays into digital signals that can be stored for subsequent
retrieval and display. A mammographer then interprets the displayed image of the breast to
determine whether the breast is normal or whether additional testing is required. This is
an alternative to traditional screening and diagnostic mammography. It can be used
whenever a traditional mammography examination is indicated.
 MAPPING
THE HEART AND TREATING ARRHYTHMIA – The NAVI-STAR®
Diagnostic/Ablation Deflectable Tip Catheter, Biosense Webster, Inc., is a
steerable, multi-electrode catheter with a deflectable tip. The catheter provides
information for electrophysiological mapping of the heart and transmits RF
(radiofrequency) current through the catheter tip electrode for ablation purposes. When
used with the CARTO® system and REF-STAR® reference device, a
real-time 3D reconstruction of the heart chamber is provided. For ablation, the catheter
is used in conjunction with a compatible RF generator and a commercially available
dispersive pad. The NAVI-STAR® catheter is available with either a
thermocouple or thermistor temperature sensor embedded in the tip electrode. A magnetic
field location sensor (location sensor) embedded in the tip transmits location information
to the CARTOsystem. Thermal energy is delivered at the site of application, which produces
a lesion that interrupts a defective electrical conduction pathway in the cardiac wall.
The device and its related accessory devices are indicated for catheter-based atrial and
ventricular cardiac mapping, and for cardiac ablation procedures
MAPPING
THE HEART AND TREATING ARRHYTHMIA – The NAVI-STAR®
Diagnostic/Ablation Deflectable Tip Catheter, Biosense Webster, Inc., is a
steerable, multi-electrode catheter with a deflectable tip. The catheter provides
information for electrophysiological mapping of the heart and transmits RF
(radiofrequency) current through the catheter tip electrode for ablation purposes. When
used with the CARTO® system and REF-STAR® reference device, a
real-time 3D reconstruction of the heart chamber is provided. For ablation, the catheter
is used in conjunction with a compatible RF generator and a commercially available
dispersive pad. The NAVI-STAR® catheter is available with either a
thermocouple or thermistor temperature sensor embedded in the tip electrode. A magnetic
field location sensor (location sensor) embedded in the tip transmits location information
to the CARTOsystem. Thermal energy is delivered at the site of application, which produces
a lesion that interrupts a defective electrical conduction pathway in the cardiac wall.
The device and its related accessory devices are indicated for catheter-based atrial and
ventricular cardiac mapping, and for cardiac ablation procedures
LASER-BASED EYE SURGERY –
The Hyperion™ LTK System, Sunrise Technologies, International, Inc., is a
new type of refractive surgical laser used in the temporary reduction of hyperopia
(farsightedness). Its benefit is temporary because the amount of farsightedness correction
decreases over time. However, some patients may retain some or all of the correction. LTK
(Laser Thermal Keratoplasty) is a surgical treatment for farsightedness performed using a
holmium YAG laser. The laser produces a beam that is positioned outside of the optical
zone of the eye. The beam heats the tissue in the cornea, causing it to shrink slightly.
When the tissue shrinks, the cornea angle becomes steeper. This allows incoming light to
focus on the retina, giving clearer images. The goal of LTK is to improve the
patient’s ability to see objects at a distance.  This device may be used to treat
patients who have farsightedness between +0.75 to +2.5 diopters (D), who are at least 40
years of age, and whose visual acuity has changed very little over time (that is, the
patient’s glasses prescription has changed no more than 0.50 diopter in the previous
six months). Treatment using this device will allow hyperopes (farsighted persons) who
have difficulty seeing clearly at a distance without glasses to have improved distance
vision without needing glasses.
This device may be used to treat
patients who have farsightedness between +0.75 to +2.5 diopters (D), who are at least 40
years of age, and whose visual acuity has changed very little over time (that is, the
patient’s glasses prescription has changed no more than 0.50 diopter in the previous
six months). Treatment using this device will allow hyperopes (farsighted persons) who
have difficulty seeing clearly at a distance without glasses to have improved distance
vision without needing glasses.
 TREATING GASTROESOPHAGEAL REFLUX DISEASE -- The
CSM Stretta System, Conway Stuart Medical, Inc., is an electrosurgical system
that includes a generator, electrosurgical catheter and a dispersive electrode. The
electrosurgical catheter has a tip with individual needle prongs, which can be placed
interstially into soft tissue to produce soft tissue coagulation. This system is intended
for the treatment of Gastroesophageal Reflux Disease (GERD). The catheter tip needles are
inserted into the soft tissue of the esophagus at the junction of the esophagus and
stomach. Controlled energy is applied for a predetermined amount of time to produce soft
tissue coagulation at the insertion site. This coagulation results in a shrinkage of the
esophageal tissue at this site resulting in a narrowing of the junction which causes a
reduction or elimination of stomach reflux of stomach acid up into the esophagus. This
treatment can be used as an alternative to the previous surgical option of fundoplication.
Fundoplication requires use of general anesthesia, a long recovery period and an extended
hospital stay since it is major abdominal surgery. The Stretta process does not require
general anesthesia and has significantly less recovery time or hospital stay time. Both of
these procedures are used only after patients have failed more conservative treatments for
GERD such as lifestyle changes, changes in diet, and use of medication to reduce
production of stomach acid. Use of this surgical procedure can, in some patients, result
in total elimination of reflux and use of GERD medications, and in other patients, surgery
can result in improved pH values showing reduced acid problems and consequently allowing
patients to use less costly or less potent GERD medications.
TREATING GASTROESOPHAGEAL REFLUX DISEASE -- The
CSM Stretta System, Conway Stuart Medical, Inc., is an electrosurgical system
that includes a generator, electrosurgical catheter and a dispersive electrode. The
electrosurgical catheter has a tip with individual needle prongs, which can be placed
interstially into soft tissue to produce soft tissue coagulation. This system is intended
for the treatment of Gastroesophageal Reflux Disease (GERD). The catheter tip needles are
inserted into the soft tissue of the esophagus at the junction of the esophagus and
stomach. Controlled energy is applied for a predetermined amount of time to produce soft
tissue coagulation at the insertion site. This coagulation results in a shrinkage of the
esophageal tissue at this site resulting in a narrowing of the junction which causes a
reduction or elimination of stomach reflux of stomach acid up into the esophagus. This
treatment can be used as an alternative to the previous surgical option of fundoplication.
Fundoplication requires use of general anesthesia, a long recovery period and an extended
hospital stay since it is major abdominal surgery. The Stretta process does not require
general anesthesia and has significantly less recovery time or hospital stay time. Both of
these procedures are used only after patients have failed more conservative treatments for
GERD such as lifestyle changes, changes in diet, and use of medication to reduce
production of stomach acid. Use of this surgical procedure can, in some patients, result
in total elimination of reflux and use of GERD medications, and in other patients, surgery
can result in improved pH values showing reduced acid problems and consequently allowing
patients to use less costly or less potent GERD medications.
Center for Devices and Radiological Health (CDRH) maintains searchable databases of devices previously approved for marketing or declared substantially equivalent to a legally marketed device at http://www.fda.gov/cdrh/mda/mda-databases.html.
The Consumer Staff in FDA’s Center for Devices and Radiological Health, Division of Small Manufacturers Assistance also provides information to consumers regarding medical devices and radiation-emitting products to enhance their ability to avoid risk, achieve maximum benefit, and make informed decisions about the use of such products.
| Website: | http://www.fda.gov/cdrh/consumer/index.html |
| E-Mail: | dsma@cdrh.fda.gov |
| Phone: | Toll Free 1-888-463-6332 or 301-827-3990 directly between
the hours of 8:00 a.m. – 4:30 p.m. EST |
The FDA Breast Implant website for consumer information is available at http://www.fda.gov/cdrh/breastimplants/index.html.
A new CDRH website entitled LASIK Eye Surgery: Learning About LASIK is available at http://www.fda.gov/cdrh/lasik/.
ODE reviews four types of marketing applications: Premarket Notification (or a 510(k) submission), Premarket Approval Application (PMA), Product Development Protocol (PDP), and Humanitarian Device Exemption (HDE). Most devices are cleared for marketing through the 510(k) process. PMAs apply to the highest risk and newly developed devices.
During Fiscal Year 2000, ODE approved 43 PMAs and 6 HDEs. These are listed below. We recommend turning to the new PMA approval website, which is available at http://www.fda.gov/cdrh/mda/index.html, for easy-to-understand one pagers for each PMA approved.
| 25-Oct-99 | P990033 | Ceramed Corp. | PEPGEN P-15 Bone Filling Augmentation Material |
| 12-Nov-99 | P980008 | LaserSight | LaserScan LSX for PRK myopia |
| 12-Nov-99 | P990014 | Bausch & Lomb Surgical, Inc. |
Hydroview Composite Hydrogel Foldable Ultraviolet (UV) -Absorbing Posterior Chamber Intraocular Lens (IOL) |
| 19-Nov-99 | P990010 | VISX | VISX Star S2 for LASIK myopia plus astigmatism |
| 03-Dec-99 | P990019 | DUSA Pharmaceuticals, Inc. |
Photodynamic Therapy |
| 03-Dec-99 | P990009 | Fusion Medical Technologies, Inc. |
Hemostatic Agent |
| 07-Dec-99 | H990007 | CryoLife, Inc. | BioGlue®Surgical Adhesive |
| 10-Dec-99 | H980006 | DataMedix Corp. | Therasphere® |
| 16-Dec-99 | P970049 | Dishler | Dishler Excimer for LASIK myopia plus astigmatism |
| 07-Jan-00 | P990016 | McCue Corp., Inc. | Ultrasonic Bone Sonometry System |
| 20-Jan-00 | P990035 | Sunlight Ultrasound Technologies, Inc. |
Ultrasonic Bone Sonometry System |
| 28-Jan-00 | P990066 | GE Medical System | Senographe 2000D (1st digital mammography) |
| 01-Feb-00 | H990011 | Nitinol Medical. Technologies, Inc |
CardioSEAL® Septal Occlusion System |
| 03-Feb-00 | P980040 | Allergan, Inc. | Sensar Soft Acrylic UV-Absorbing Posterior Chamber IOL |
| 23-Feb-00 | P990027 | Bausch & Lomb | Technolas 217A for LASIK myopia |
| 24-Feb-00 | P990023 | Alcon Laboratories | Cellugel Ophthalmic Viscosurgical Device |
| 09-Mar-00 | H990008 | Interpore Cross International |
Telescopic Plate Spacer (TPS) Spinal System |
| 17-Mar-00 | P990054 | Cardiac Pathways Corp. | CHILLI COOLED ABLATION SYSTEM with Tracking |
| 31-Mar-00 | H990014 | Medtronic, Inc. | Gastric Electrical Stimulation System (Now known as Enterraä Therapy System) |
| 02-Apr-00 | P990013 | Star Surgical, Co. | IOL |
| 12-Apr-00 | P990048 | Carl Zeiss | VISULAS 690s Laser and VISULINK PDT adapter |
| 12-Apr-00 | P990049 | Coherent Medical Group | Coherent Opal Photoactivator and modified Coherent LaserLink |
| 18-Apr-00 | P950020 | Interventional Technologies, Inc. | Cutting Balloon™ |
| 10-May-00 | P990074 | McGhan Medical Corp. | RTV Saline-Filled Breast Implant |
| 10-May-00 | P990075 | Mentor Corporation | Saline-Filled and Spectrum® Mammary Prosthesis |
| 11-May-00 | H990012 | Cardiovascular Diagnostics, Inc. | TAS Ecarin Clotting Time Test |
| 12-May-00 | P990053 | Nellcor Puritan Bennett, Inc. | Oxifirst™ Fetal Oxygen Saturation Monitor |
| 26-May-00 | P990028 | Focal Inc. | FocalSeal-L Synthetic Absorbable Surgical Sealant |
| 31-May-00 | P990071 | Biosense Webster, Inc. | Stockert 70 RF Generator for Cardiac Ablation |
| 13-Jun-00 | P990030 | Cohesion Technologies, Inc. | CoStasis Surgical Hemostar |
| 14-Jun-00 | P980050 | Medtronic, Inc. | Medtronic® Jewel® AF 7250 Dual Chamber Implantable Cardioverter Defibrillator, Model 9961 Programmer Application Software and Medtronic® Sprint™ Model 6943 Steroid Eluting, Screw-in, Atrial/Ventricular Lead |
| 15-Jun-00 | P990025 | Biosense Webster, Inc. | NAVI-STAR® Diagnostic/Ablation Deflectable Tip Catheter |
| 22-Jun-00 | P990037 | Vascular Solutions, Inc | Vascular Solutions Duett™ Sealing Device |
| 30-Jun-00 | P990021 | QLT Photo Therapeutics, Inc. | Diomed 630 PDT Laser, Model T |
| 30-Jun-00 | P990078 | Sunrise | Hyperion LTK for hyperopia |
| 11-Jul-00 | P990018 | Menicon U.S.A. | Minicon™ (tisilfocon A) Rigid |
| 14-Jul-00 | P990064 | Medtronic, Inc. | MOSAIC® Porcine Bioprosthesis, Models 301 and 310 |
| 21-Jul-00 | P990034 | Medtronic, Inc. | Medtronic Isomed Infusion System |
| 24-Jul-00 | P000006 | Mentor Corp. | Alpha I Inflatable Penile Prosthesis |
| 01-Aug-00 | P990039 | Metra Biosystems | QUS-2 Calcaneal Ultrasonometer |
| 22-Aug-00 | P990072 | Westcon Contact Lens Co., Inc. | Horizon 55 EW and Horizon 55 |
| 31-Aug-00 | P990052 | Symphonix Devices, Inc. | Vibrant P Soundbridge System |
| 05-Sep-00 | P970042 | Medstone International, Inc. | Medstone STS™ Lithotripter |
| 08-Sep-00 | P990055 | Bayer Corp. | Bayer Immuno 1 Complexed PSA Assay |
| 19-Sep-00 | P980010 | Ostenometer MediTech, Inc. | DTU-one Ultrasound Scanner |
| 25-Sep-00 | P990040 | Cordis Neuro- vascular Inc. | TRUFILL®n-Butyl Cyanoacrylate (nBCA) Liquid Embolic System |
| 29-Sep-00 | P000014 | Ortho-Clinical Diagnostics, Inc. | VITROS Immunodiagnostic Products:Anti-HBS Reagent Pack/Anti-HBS Calibrators |
| 29-Sep-00 | P000009 | Biotronik, Inc. | Phylax AV Implantable Cardioverter Defibrillator with Program Software (I-GAV.2.U) |
| 29-Sep-00 | P000011 | Biocompatibles Cardiovascular, Inc. | BiodivYsio™ AS PC (phosphorylcholine) Coated
Stent and Delivery System |
The following devices were approved via PMAs, PMA Supplements, and HDEs or cleared via 510(k)s or classified via the Automatic Evaluation of Class III Designation process during FY 00. They represent significant medical breakthroughs because they are first-of-a-kind, e.g., they use a new technology or energy source, or they provide a major diagnostic or therapeutic advancement, such as reducing hospital stays, replacing the need for surgical intervention, reducing the time needed for a diagnostic determination, etc. The information for each device includes the trade name and/or classification name, firm, and date of approval or clearance.
Medtronic® Jewel® AF 7250 Dual Chamber Implantable Cardioverter Defibrillator by Medtronic, Inc. (June 14, 2000)
NAVI-STAR® Diagnostic/Ablation Deflectable Tip Catheter by Biosense Webster, Inc. (June 15, 2000)
TAS Ecarin Clotting Time Test by Cardiovascular Diagnostics, Inc. (May 11, 2000)
RTV Saline-Filled Breast Implant by McGhan Medical Corp. (May 10, 2000)
Saline-Filled and Spectrum® Mammary Prosthesis by Mentor Corporation (May 10, 2000)
FocalSeal-L Synthetic Absorbable Surgical Sealant by Focal Inc. (May 26, 2000)
Apligraf® (Graftskin) by Organogenesis Inc. (June 20, 2000)
TRUFILL® n-Butyl Cyanoacrylate (n-BCA) Liquid Embolic System by Cordis Neurovascular Inc. (September 25, 2000)
Hyperion® for Laser Thermal Keratoplasty for Hyperopia (+0.75 to +2.5 diopters) by Sunrise Technologies (June 30, 2000)
Vibrant Soundbridge by Symphonix Devices, Inc. (August 31, 2000)
Senographe 2000D (1st digital mammography) by GE Medical System (January 28, 2000)
Gastric Electrical Stimulation System by Medtronic, Inc. (March 31, 2000)
Oxifirst™ Fetal Oxygen Saturation Monitor by Nellcor Puritan Bennett, Inc. (May 12, 2000)
Alpha I Inflatable Penile Prosthesis by Mentor Corp. (July 14, 2000)
Medstone STS™ Lithotripter by Medstone International, Inc. (September 5, 2000)
510(k) Clearances or Automatic Evaluations of Class III Designation Devices (AE)
Becton, Dickinson & Co.’s Probetec ET System for Chlamydia Trachomatis and Gonorrhea (November 4, 1999)
Wallac Neonatal Biotinidase Test Kit by Perkin Elmer Inc. (November 22, 1999)
Axix %CDT Turbidometric Immunoassay by Axis (December 21, 1999)
MTM Bioscanner HDL Test Strips (Over-the-Counter) by Polymer Technology Systems, Inc. (January 13, 2000)
CDC’s Synthetic VDRL Antigen Slide for Syphilis (February 23, 2000)
Cedia Dau Amphassure Assay by Microgenics Corporation (May 2, 2000)
BV Blue by Gryphus Diagnostics, L.L.C. (May 15, 2000)
Bioscanner Triglycerides Test Strips by Polymer Technology Systems, Inc. (May 24, 2000)
Microwave Delivery System (MDS), Model MMC-3000 by Microwave Medical, Inc. (October 1, 1999)
600 C Laser Keratome by IntraLase Corporation (December 17, 1999)
Excimer Laser Phototherapy System AL7000 by AccuLase, Inc. (January 27, 2000)
Visage Cosmetic Surgery Model V5000 by ArthroCare Corporation (March 20, 2000)
CSM Stretta System by Conway Stuart Medical Inc. (April 18, 2000)
Laser Photolysis System and Pharo Opthalmic Surgery System by A.R.C. Laser Corporation (June 29, 2000)
da Vinci™ Endoscopic Instrument Control System and Endoscopic Instruments by Intuitive Surgical, Inc. (July 11, 2000)
Purilens System contact lens cleaning and disinfection system by Purilens, Inc. (October 1, 1999)
Ocu-flex-38 Keratoconus (polymacon) soft contact lens by Ocu-Ease Optical Products, (October 4, 1999)
Hylashield CL contact lens lubricating eye drop by Biomatrix, Inc. (March 2, 2000)
Hylasine, hylan B Gel by Biomatrix, Inc. (March 13, 2000)
VISX WaveScan Wavefront Analysis System Refractometer (April 28, 2000)
Autononmous Technologies CustomCornea Wavefront Analysis Refractometer (May 16, 2000)
Sportsight GP rigid gas permeable contact lens by Paragon Vision Sciences, (May 22, 2000)
Option care system for cleaning and disinfecting non-UV absorbing contact lenses by Optisonic, Inc. (June 5, 2000)
MeroGel Otologic pack by Medtronic Xomed (July 3, 2000)
LifeSite® Hemodialysis Access System by Vasca, Inc. (August 24, 2000)
The following guidance documents were adopted by ODE and its operating divisions during FY 00 and are available from the Division of Small Manufacturers Assistance (DSMA, HFZ-200). To contact DSMA, call 800-638-2041 or 301-443-6597; fax 301-443-8818; Email dsma@cdrh.fda.gov or write to DSMA (HFZ-200, Food and Drug Administration, 1350 Piccard Drive, Rockville, Maryland 20850-4307.)
Many are also available through the CDRH Facts-On-Demand (faxback service at 800-899-0381 or 301-837-0111) and the World Wide Web (CDRH homepage: http://www.fda.gov/cdrh) which provide easy access to the latest information and operating policies and procedures.
ODE
Use of Standards in Substantial Equivalence Determinations (March 13, 2000)
Guidance for Over-the-Counter (OTC) Human Chorionic Gonadotropin (hCG) 510(k)s (July 22, 2000)
Guidance for Over-the-Counter (OTC) Ovulation Predictor 510(k)s (July 22, 2000)
Class II Special Control Guidance Document for Anti-Saccharomyces cerevisia (S. cerevisiae) Antibody (ASCA) Premarket Notification (August 23, 2000)
DCRD
Guidance for Cardiovascular Intravascular Filter Submissions (November 26, 1999)
Guidance for Annuloplasty Rings 510(k) Submissions (November 26,1999)
Guidance for Premarket Notification Submissions for Nitric Oxide Delivery Apparatus, Nitric Oxide Analyzer and Nitrogen Dioxide Analyzer (January 24, 2000)
Guidance for Indwelling Blood Gas Analyzer 510(k) Submissions (February 21, 2000)
Guidance for Electrical Safety, Electromagnetic Compatibility, Mechanical Testing for Indwelling Blood Gas Analyzer Premarket Notification Submissions (June 28, 2000)
Class II Special Control Guidance for Acute Upper Airway Obstruction Devices (July 3, 2000)
One Consolidated Annual Report for a Device Product Line (1-CARD): Pilot for Preparation of Annual Reports for Pacemaker Premarket Approval Applications (July 6, 2000)
Draft Guidance for Infant/Child Apnea Monitor 510(k) Submissions (September 22, 2000)
DGRND
Guidance on Preclinical and Clinical Data and Labeling for Breast Prostheses (October 5, 1999)
Guidance for Resorbable Adhesion Barrier Devices for Use in Abdominal and/or Pelvic Surgery (December 16, 1999)
Guidance Document for the Preparation of IDEs for Spinal Systems (January 13, 2000)
Guidance for Surgical Suture 510(k)s (August 10, 2000)
Guidance for Spinal System 510(k)s (September 27, 2000)
DOED
Intraocular Lens Guidance Document (draft) (October 14, 1999)
Guidance for Premarket Submissions of Orthokeratology Rigid Gas Permeable Contact Lenses (April 10, 2000)
Refractive Implants: Guidance for Investigational Device Exemptions (IDE) and Premarket Approval (PMA) Applications (draft) (August 1, 2000)
DRARD
Draft Guidance for Resorbable Adhesion Barrier Devices for Use in Abdominal and/or Pelvic Surgery (December 16, 1999)
Guidance for the Content of Premarket Notifications for Penile Rigidity Implants; Final (January 16, 2000)
Class II Special Controls Guidance Document for Clinical Engorgement Devices (July 3, 2000)
Guidance for the Submission of Premarket Notifications for Medical Image Management Devices (July 27, 2000) (update of PACS guidance, DSMA FOD#416)
Guidance for the Submission of Premarket Notifications for Photon-Emitting Brachytherapy Sources (August 2, 2000)
Guidance on Premarket Approval Applications for Assays Pertaining to Hepatitis C Viruses (HCV) that are Indicated for Diagnosis or Monitoring of HCV Infection or Associated Disease (October 8, 1999)
Guidance on the Labeling for Over-the-Counter Sample Collection Systems for Drugs of Abuse Testing (December 21, 1999)
Guidance on Review Criteria for Assessment of Antimicrobial Susceptibility Devices (March 8, 2000)
Revision to the Extracorporeal Shockwave Lithotripter Guidance (August 9, 2000)
Guidance for Administrative Procedures for CLIA Categorization (August 14, 2000)
ODE is responsible for protecting the rights, safety and welfare of patients participating in clinical studies of significant risk medical device research and for evaluating the safety and effectiveness of medical devices before these devices enter the U.S. market place.
Following are the details of ODE’s review activities and performance for Fiscal Year 2000 (FY 00). Most of the data below can be found in the tables in Part 5 -- the Operational Summary section of this report. First, we present the major submissions received and completed. Next, we review the Premarket Approval Applications (PMAs) in terms of review time as well as volume. This same analysis is done for PMA supplements. The remainder of this section deals with Humanitarian Device Exemptions (HDEs), Investigational Device Exemptions (IDEs), and Premarket Notifications (510(k)s).
During FY 00, ODE received a total of 16,919 submissions, compared to 16,812 in FY 99; 9,774 were major submissions compared to 9,792 last fiscal year [see Table 1]. Major submissions include: IDEs -- originals, amendments and supplements; PMAs -- originals and supplements; HDEs -- originals and supplements; and 510(k)s. Other submissions include PMA amendments and reports; master files; and 510(k) amendments and supplements.
Table 1. Major
Submissions Received
FY 90 – FY 00
| Type of Submission |
|
|
|
|
|
|
|
|
|
|
|
Orig. PMAs |
79 |
75 |
65 |
40 |
43 |
39 |
44 |
66 |
47 |
60 |
67 |
| PMA Supp. | 660 | 593 | 606 | 395 | 372 | 499 | 415 | 409 | 513 | 552 | 545 |
| Orig. IDEs | 252 | 213 | 229 | 241 | 171 | 214 | 253 | 297 | 322 | 304 | 311 |
| IDE Amend. | 288 |
283 |
297 |
320 |
254 |
210 |
219 |
223 |
226 |
275 |
240 |
| IDE Supp. | 3,043 | 3,647 | 3,644 | 3,668 | 3,020 | 3,171 | 3,189 | 3,776 | 4,277 | 4,127 | 4,388 |
| 510(k)s | 5,831 | 5,770 | 6,509 | 6,288 | 6,434 | 6,056 | 5,297 | 5,049 | 4,623 | 4,458 | 4,202 |
| Orig. HDE | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 8 | 12 | 11 |
| HDE Supp. | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 10 |
| _____ | _____ | _____ | _____ | _____ | _____ | _____ | _____ | _____ | _____ | _____ | |
| Total | 10,153 | 10,581 | 11,350 | 10,952 | 10,293 | 10,189 | 9,417 | 9,824 | 10,016 | 9,792 | 9,774 |
On the decision side, ODE completed the processing of 9,994 major submissions, compared to 9,881 major submissions in FY 99. [See Table 2 for major submissions completed.]
Table 2. Major
Submissions Completed
FY 90 - FY 00
| Type of Submission |
|
|
|
|
|
|
|
|
|
|
|
Orig. PMAs |
47 |
27 |
12 |
24 |
26 |
27 |
43 |
48 |
46 |
45 |
43 |
| PMA Supp. | 700 | 479 | 394 | 354 | 385 | 435 | 462 | 401 | 421 | 437 | 474 |
| Orig. IDEs | 248 | 220 | 215 | 248 | 174 | 210 | 260 | 272 | 325 | 305 | 320 |
| IDE Amend. | 270 |
287 |
297 |
324 |
256 |
213 |
218 |
220 |
225 |
268 |
251 |
| IDE Supp. | 2,968 | 3,705 | 3,469 | 3,814 | 3,070 | 3,181 | 3,121 | 3,777 | 4,209 | 4,224 | 4,335 |
| 510(k)s | 6,197 | 5,367 | 4,862 | 5,073 | 7,135 | 7,948 | 5,563 | 5,155 | 5,229 | 4,593 | 4,397 |
| Orig. HDE | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 4 | 6 | 6 |
| HDE Supp. | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 10 |
| _____ | _____ | _____ | _____ | _____ | _____ | _____ | _____ | _____ | _____ | _____ | |
| Total | 10,430 | 10,085 | 9,249 | 9,837 | 11,045 | 12,014 | 9,667 | 9,875 | 10,459 | 9,881 | 9,994 |
ODE ended the fiscal year with 359 employees. During the year, 27 full-time employees (12 scientific reviewers, 1 medical officer, 13 clericals and 1 program analyst) left through resignation or retirement. During FY 00, 52 new employees (30 scientific reviewers, 6 medical officers, 1 program analyst, 11 clericals, and 4 summer students) joined our office.
Premarket Approval Applications (PMAs)
[NOTE: In previous annual reports, the PMA data included data for Humanitarian Device Exemption (HDE) Applications. This annual report contains a separate section for HDEs (see page 19). We also added new statistical Tables 8, 9 and 10 that contain HDE data.]
ODE received 67 complete original PMAs (7 more than the number received in FY 99) and 55 modular submissions representing 48 PMA shells.
The total number of PMAs in inventory (active and on hold) at the end of this fiscal year decreased from 77 in FY 99 to 76. The number of active PMAs under review decreased at the end of FY 00 to 35 compared to 47 last year, and those on hold increased from 30 in FY 99 to 41 in FY 00. This means that we took action on more PMAs and thus reduced the number under active review. For the third consecutive year, there were no active and overdue PMAs at the end of the fiscal year.
The total number of PMA actions increased from 229 to 321 actions. These actions included 68 filing decisions, 173 review determinations, and 80 approval/approvable/not approvable decisions.
The 80 original PMA decisions were comprised of 43 approved PMAs, 33 approvable PMAs, and 4 not approvable PMAs. None of the 43 approvals were expedited PMAs. See Part 2 (INDUSTRY INFORMATION) for a complete list of PMA approvals.
Average FDA review time for original PMAs reaching approval increased from 149 days in FY 99 to 158 days in FY 00. The non-FDA component of review time increased from 26 days in FY 99 to 40 days this fiscal year. Thus, the total average review time increased to 198 days. Of greater significance to industry is the total elapsed time from submission to decision.
Figure 1. Average Review Time for PMA Decision Cohort Approvals
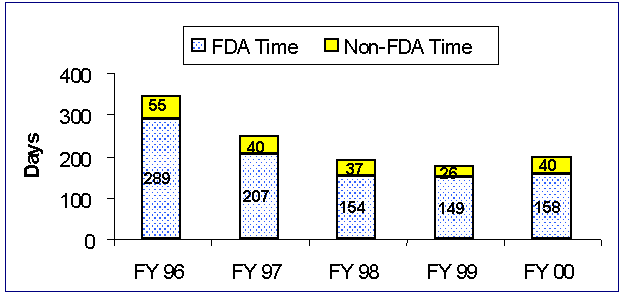
In FY 00, the total average elapsed time for PMA decision cohort performance decreased from 380 days in FY 99 to 362 days in FY 00. (Please refer to Table 4.)
Figure 2. Original Receipt Cohort PMAs Received and Filed
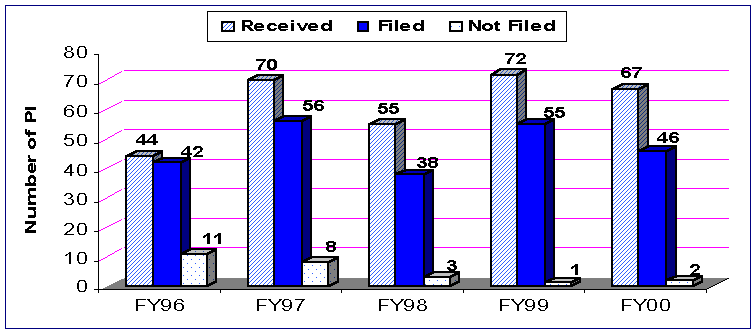
Figure 3. Receipt
Cohort PMA Average Elapsed
Time from Filing to Final Action
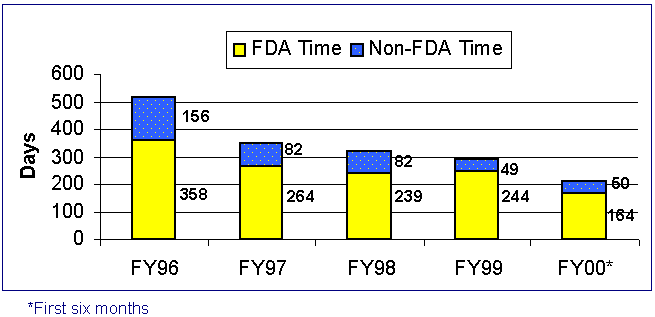
For the first 6 months of FY 00 for PMA receipt cohort performance, the average FDA days from filing to first action decreased from 145 in FY 99 to 139 days.
The average FDA (total) elapsed time to an approval or to a denial decreased from 244(293) in FY 99 to 164(214) days in FY 00. The median FDA (total) elapsed time to an approval or denial decision decreased from 240(269) in FY 99 to 178(211) days in FY 00. This means that all of the statistics of the PMA receipt cohort for FY 00 indicate that we are making decisions faster.
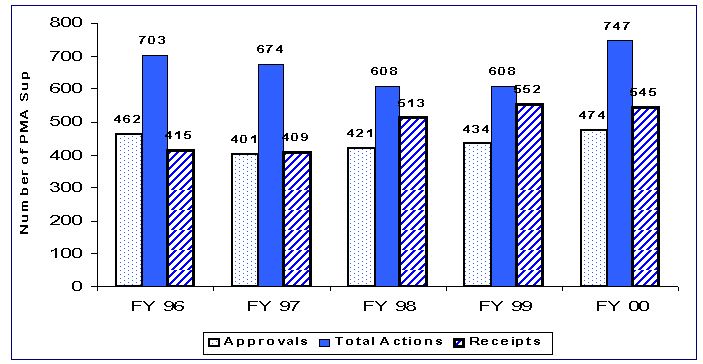
The number of PMA supplements received decreased from last year’s 552 to 545. There were 747 PMA supplement actions which is up from last year’s 608 total actions. These actions included 17 panel track PMA supplement filing decisions, 98 scientific review decisions, and 632 approval decisions.
Figure 4. Annual Receipts and Actions for PMA Supplement Decision Cohort
For PMA supplements reaching final action, the average elapsed FDA review time increased from 92 days in FY 99 to 94 days in FY 00, and the total average elapsed time increased from 118 days to 122 days.
Figure 5. Average Review Time for PMA Supplements
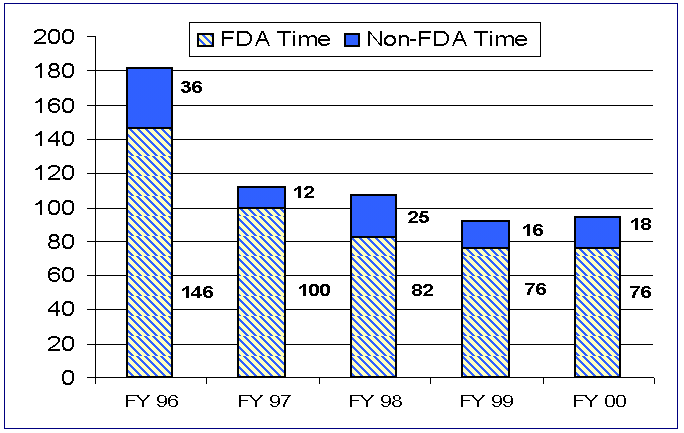
Just as in FY 97, FY 98 and FY 99, there were no PMA supplements active and overdue at the end of this fiscal year. The number of active supplements decreased to 98 in FY 00 from 158 in FY 99, while the number of supplements on hold increased from 69 to 84. This means that although we are receiving about the same number of PMA supplements, we are reaching final decisions on more, but we are taking an average of 3 extra days for the decisions.
For the first 6 months of FY 00 for PMA supplements receipt cohort performance, the first action and final action as follows. The average FDA days from filing to first decision decreased from 74 in FY 99 to 67 days in FY 00. The average FDA (total) elapsed time to an approval or denial decreased from 79(95) in FY 99 to 66(76) in FY 00. The median FDA (total) elapsed time to an approval or denial remained the same from 35(47) in FY 99 to 35(43) days in FY 00.
Real-Time Review of PMA Supplements
A total of 146 requests were received and processed for real time PMA supplements in FY 00 which represents 27% of all supplements received. Of those submissions, 134 were approved. Most applicants chose telephone conferencing versus a face-to-face meeting or a videoconference. The majority of these applications were reviewed in DCRND (54%) followed by DGRD (23%), DOD (11%), DRAERD (8%), DDIGD (2%) and DCLD (1%). Overall, average review time from "receipt" to first action (approvable, not approvable or approval order) was 34 days, and was 38 days from receipt to approval.
Product Development Protocols (PDPs)
Two PDPs have been approved in FY 00, and reports are being received on their progress for the clinical study. No original Notices of Completion were declared complete. In addition, five "Real Time" supplements, and three routine PDP supplements were approved. Note that a PDP that has been declared complete is considered to have an approved PMA. ODE continues to encourage the use of the PDP process and will work with the interested applicants to fully evaluate their PMA options.
ODE received a total of 48 PMA shells and 55 modules. A total of 17 modules were found to be acceptable while 12 received deficiency letters. A number of modules were rolled into PMA review during FY 00 because they were under review or on hold at the time the PMA was received. Applicants with modular submissions that were under review or deficient when the PMA was received continued to receive feedback under the PMA for those modules. Review times for PMAs that had modular submissions were approximately half that for traditional PMAs. However, this is based on a small number of submissions achieving PMA approval since modular review was implemented. A tracking system with modular PMA query capability became available during FY 99.
Humanitarian Device Exemption (HDE) Applications
ODE received 11 original HDEs, 1 less than the number received in FY 99. The total number of original HDE actions decreased from 37 in FY 99 to 36 in FY 00. These actions included 12 filing decisions, 16 review determinations, 7 approval decisions and 1 other final decision.
A total of 8 first actions were made this fiscal year, a decrease from 13 made last year. The average time from filing to first action decreased from 87 days in FY 99 to 61 days in FY 00.
One hundred percent of the first actions made in FY 00 occurred within 75 days.
The 7 approval decisions were comprised of 6 approved HDEs and 1 approvable HDE.
In FY 00, the average elapsed time (from filing to final approval) for original HDEs was 216 days, an increase from 163 days in FY 99. The average FDA time was 112 days, a decrease from 113 days in FY 99. The average non-FDA time was 104 days, a significant increase from 50 days last year.
The total number of original HDEs in inventory (active and on hold) at the end of this fiscal year was 10, the same as last fiscal year. Of these, 2 were under review and 8 were on hold. There was no active HDEs that were overdue at the end of the fiscal year.
The number of HDE supplements received increased from 4 in FY 99 to 10 in FY 00. There were 11 HDE supplement actions in FY 00, up from 7 in FY 99. These actions included 10 approval decisions and 1 not approvable decision.
A total of 10 first actions for HDE supplements were made this fiscal year, an increase from 4 last year. The average time from filing to first action decrease from 57 days in FY 99 to 44 days in FY 00. One hundred percent of the first actions were made within 75 days.
The average elapsed time (from filing to final approval) for HDE supplements decreased from 94 days in FY 99 to 76 days in FY 00. The average FDA time decreased from 70 days in FY 99 to 43 days in FY 00. Non-FDA time increased from 24 days in FY 99 to 33 days in FY 00.
The number of HDE supplements in inventory (active and on hold) at the end of this fiscal year was 1, the same as last fiscal year.
Investigational Device Exemptions (IDE)
During FY 00, ODE reviewed 244 pre-IDEs. Based on these reviews, guidance for the pre-original IDE submissions were provided through meetings with the sponsors, letters, fax, or by phone phone.
ODE received 311 original IDEs, an increase from 304 received in FY 99. There were 320 decisions made on original IDEs, an increase from 305 last year.
Ninety-nine percent of all original IDE decisions were issued within 30 days in FY 00. The average review time was 28 days.
Figure 6. Percentage of IDEs Approved on First Review Cycle*
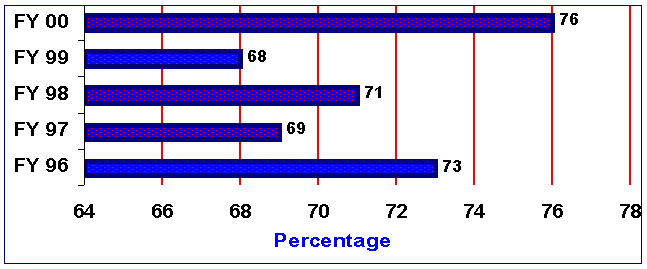
*Based on those IDEs complete enough to permit substantial review.
Of the IDEs which were complete enough to support substantive review, the percentage of IDEs approved on the first review cycle increased from 68% in FY 99 to 76% in FY 00.
During this fiscal year, 240 IDE amendments were received. Decisions were made on 251 amendments: 107 approvals (43%); 34 disapprovals (13%); and 110 other administrative actions (44%). One hundred percent of these decisions were made within 30 days.
It took an average total time of 136 days to approve IDEs that were initially disapproved, down from 145 days in FY 99. This average approval time consisted of 70 days for FDA time, up from 57 days last year, and 66 days for non-FDA time, down from 88 days in FY 99.
ODE received 4,388 IDE supplements during FY 00. There were no overdue supplements at the end of the year, and the percentage of supplements reviewed within the 30-day statutory timeframe was 100 percent in FY 00. The average review time for IDE supplements stayed the same at 20 days.
Premarket Notification (510(k)s)
ODE received 4,202 original 510(k)s, as well as 1,742 510(k) supplements (responses to hold letters, the receipt of which restart the 90-day review clock), and 2,953 510(k) amendments (additional information received while the 510(k) is under review, the receipt of which does not affect the review clock).
The total average review time remained at 102 days in FY 00, and the average FDA review time was 77 days, down from 80 days in FY 99. The median review time, i.e., the time it took to review 50% of the 510(k)s, has been falling from a high of 164 days in FY 93 to a current low of 72 days in FY 00.
Figure 7. Average 510(k) Review Time for Decision Cohort
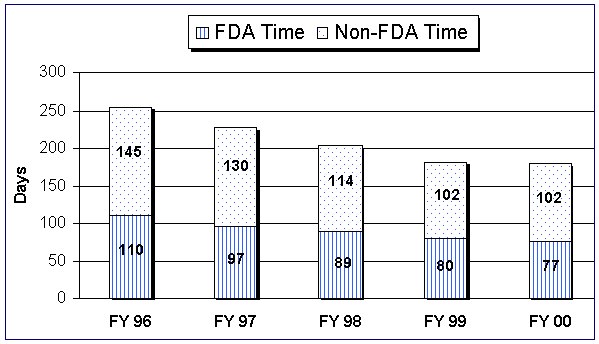
There were 1,220 510(k)s in inventory (those under active review or on hold) at the end of this fiscal year, which is 184 less than the 1,404 in FY 99’s end-of-year inventory. The number on hold decreased from 461 at the end of FY 99 to 370. Most important, for the fifth consecutive fiscal year there was no 510(k)s active and overdue at the end of the reporting period.
For the first 9 months of FY 00 for receipt cohort performance, the FDA time from receipt to final decision decreased to 60 days compared to 66 days for the first 9 months in FY 99.
Figure 8. Receipts and Actions for 510(k) Receipt Cohorts*
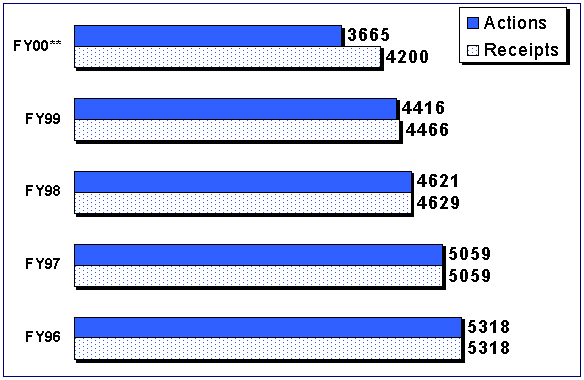
*Cut off Date of 8/30/00 for all receipt cohorts.
**12 months projection based on first 9 months of receipts.
For the first 9 months of FY 00 for receipt cohort performance, the total time from receipt to final decision decreased to 75 days compared to 77 days for the first 9 months in FY 99.
Figure 9. FDA Days from Receipt to Final Action for 510(k) Receipt Cohorts*
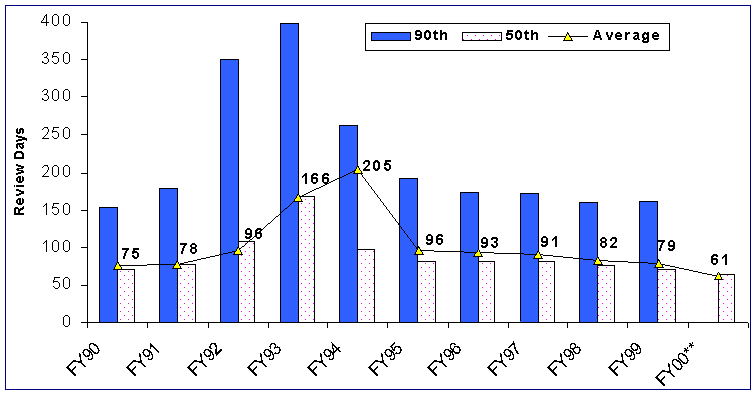
*Cut Off Date as of 9/30/00 for all
receipt cohorts.
**For the first 9 months of FY 00. 90th percentile data not available for FY 00.
During FY 00, ODE received 47 510(k)s reviewed by third-party organizations under the Accredited Persons provisions (section 523) of the Federal Food, Drug, and Cosmetic Act. This is a small percentage of all 510(k)s that were eligible for third-party review, but is a 47% increase over the number of such submissions received by ODE last fiscal year. ODE made final decisions on 46 "third-party" 510(k)s in FY 00, an increase from the 29 final decisions in FY 99. The average total elapsed time from a third party’s receipt of a 510(k) to ODE’s issuance of a substantial equivalence decision was 68 days, as compared to the average total elapsed time of 99 days for ODE’s decision on comparable 510(k)s that did not have a third-party review.
In June 2000, to encourage greater industry use of Accredited Persons, the Center expanded the list of Class I and Class II devices that are eligible for review from 154 devices to 211 devices. In the Federal Register on July 18, 2000 (65 FR 44540), the Center also proposed an expansion pilot that would permit third-party review of a greatly expanded list of devices. The pilot would allow-subject to certain specified conditions-third-party review of Class II devices for which device-specific guidance does not exist.
Until now, device-specific guidance had existed for each Class II device that is eligible for third-party review. The Federal Register notice established a 45-day public comment period, which ended September 1, 2000. The Center has reviewed the public comments and intends to finalize the proposal in FY 01. Information on the expansion pilot is available on the Center’s third party web page at http://www.fda.gov/cdrh/thirdparty.
From October 1, 1999 to September 30, 2000 ODE received 615 Special 510(k)s out of the 4,202 total number of 510(k)s received, and 583 have received final decisions with the average FDA review time of 27 days and the average total time of 32 days, and 551 were found substantially equivalent and the remaining 32 had other decisions such as withdrawn or deleted.
During the same timeframe ODE received 150 Abbreviated 510(k)s out of the 4,202 total number of 510(k)s received. One hundred eighteen received final decisions (104 substantially equivalent and 12 other decisions, and 2 NSEs) with a FDA average review time of 83 days and total time of 103 days. None of the Abbreviated 510(k)s went over 90 days.
Significant Medical Device Breakthroughs
During FY 00, ODE approved 15 PMAs and cleared 25 510(k)s that represented significant medical device breakthroughs. See INDUSTRY INFORMATION for a complete listing.
Automatic Evaluation of Class III Designation
Final Reclassification Actions
Guidance for Industry and Reviewers
In FY 00, ODE published 25 final guidance documents and published 5 draft guidance documents for comment. See INDUSTRY INFORMATION for a complete listing of all ODE guidance documents published in FY 00.
The FDA Modernization Act of 1997 contains a charge to FDA to require only clinical data or information necessary to establish device effectiveness or to confer substantial equivalence. FDA must consider the least burdensome means of demonstrating effectiveness or equivalence in the review of premarket applications. Pursuant to this congressional mandate, ODE has taken the lead in implementing the concept of "least burdensome." As part of this effort, ODE actively participated in a CDRH-wide working group on least burdensome issues. As part of the efforts to implement the least burdensome provisions of FDAMA, ODE's internal tracking documents and correspondence with companies have been modified to highlight least burdensome efforts. Working collaboratively with an Industry Task Force, ODE participated in the preparation of a draft "Concepts and Principles" document. Efforts are continuing, both internally and with the Industry Task Force, to implement the least burdensome provisions in all of our activities. Information related to the least burdensome provisions of the FDA Modernization Act of 1997 can be accessed on the CDRH website: http://www.fda.gov/cdrh/modact/leastburdensome.html.
Significant Jurisdictional Issues Involving Devices in FY 2000
Title 21 of the Code of Federal Regulations Part 3 - PRODUCT JURISDICTION describes the procedure the agency uses to assign Center jurisdiction over medical products whose jurisdiction is not clear or is in dispute. Requests for Designations (RFDs) over such products are made in writing to the FDA Office of the Chief Mediator and Ombudsman. These formal submissions contain the material describing the requester's product and their proposal regarding which Center should be given the lead designation over the product and whose authorities (Biological, Device or Drug) should apply.
In FY 2000, CDRH participated in the review of 21 out of 23 RFDs (two were assigned wholly to CDER and CBER only) in addition to completing the review of 2 RFDs received in FY 99. The reviews of the 21 new requests were assigned to the ODE Divisions as follows; DGRND was assigned to review seven and shared an additional review with DOED, DDIGD and DCRD were assigned five each, DOED was assigned one and shared one with DGRND, and DCLD was assigned one RFD. Finally, one was not assigned to any division as it was handled by the Center’s coordinator for incoming RFDs.
Out of the 21 RFDs assigned to CDRH for review, seven were not due for completion until FY 2001. Of the 16 RFD’s whose reviews were completed, CDRH was assigned the lead center in 10 of those requests and one was withdrawn before its review could be completed. Of the remaining five the lead center designation was to either CDER or CBER.
The Center's Medical Devices Advisory Committee (MDAC) provides advice to FDA on the safety and effectiveness of marketed and investigational devices, the classification of devices into one of three regulatory categories, the possible risks to health associated with the use of devices, the formulation of product development protocols, the review of premarket approval applications, and the content of guidelines or guidance documents designed to improve the interaction between the Agency and sponsors of medical devices. The MDAC consists of 18 panels divided according to medical device specialties.
In FY 00, ODE held 27 panel meetings, 12 open meetings and 15 partially closed. Of the 18 panels, four met at least once, four met twice, and five met three times during the fiscal year. The panels collectively considered 28 PMA submissions, five PMA Supplements, four Reclassifications, two PDPs, one 510k, and two guidance documents. The panels discussed and provided advice on a number of issues. Topics ranged, for example, from the design of clinical trials to support claims for reduced posterior capsular opacification for intraocular lenses, devices used in atrial fibrillation therapies, to assessing the performance of in vitro diagnostic tests for hepatitis infection. Further information about government-wide advisory committees is available at the Federal Advisory Committee Act Database on the GSA website: http://www.gsa.gov/Portal/policy.jsp?detail=longDesc&OID=116001.
There were 25 formal training sessions for new panel members (special government employees known as SGEs). The two-hour training for SGEs covered the laws and regulations with respect to medical devices, organizational structure of the Agency, ODE's operations, the roles and responsibilities of panel members, the elements of a panel meeting, and conflict of interest.
Panel members are leading authorities in a broad range of medical areas and have current experience in medical practice, teaching and/or research. Each panel has a consumer representative, an industry representative, and when appropriate, a patient representative; these panel members do not vote but provide valuable input into panel discussions. Patient representatives served on two panels during the fiscal year – the Clinical Chemistry and Clinical Toxicology Devices Panel meeting on December 6 and 7, 1999, and the Neurological Devices Panel on March 31, 2000. During the past fiscal year, females made up 43% of the ODE panel membership and minorities approximately 31%.
ODE continuously recruits highly qualified experts to serve as consultants and panel members. Potential candidates are asked to provide detailed information concerning financial holdings, employment, and research grants and contracts to identify any potential conflict of interest. Interested individuals should send their resume to the Advisory Panel Coordinator, Office of Device Evaluation, 9200 Corporate Boulevard, Rockville, Maryland 20850.
Announcements of panel meetings are publicized in several ways: voice information via the FDA Advisory Committee Information Line (1-800-741-8138), printed information in the Federal Register, and on the Internet (http://www.fda.gov/cdrh/panelmtg.html). This website also includes summaries of the most recent advisory panel meetings.
The Guidance on Amended Procedures for Advisory Panel Meetings was revised on July 22, 2000, to clarify the standard operating procedures that apply to advisory panel meetings where a specific submission is being considered by the panel or to device classification panel meetings on issues involving more than one sponsor. The clarification addresses timeframes for when and what types of information/new data analyses might be submitted to the panel. The revised guidance is available at http://www.fda.gov/cdrh/modact/amendpan.html.
During this fiscal year, ODE investigated about 62 cases concerning the integrity of data submitted to the agency in premarket applications. Under the Application Integrity Program (AIP), two firms were placed on the AIP list and AIP restrictions applied against these firms.
ODE handled 37 instances related to questions arising under the standards of conduct for employees. During FY 00, as in years past, the ODE staff received several unsolicited gifts from the regulated industry. Both the offering of gifts and their acceptance in general, are prohibited under applicable laws and regulations. The regulated industry, their agents and representatives should not send gifts to staff members. (See Standards of Ethical Conduct for Employees of the Executive Branch on the internet at http://www.usoge.gov/pages/forms_pubs_otherdocs/fpo_files/reference/rfsoc_99.pdf).
Freedom of Information Requests
ODE staff received 1,080 FOI requests during FY 00, a decrease from 1,355 last fiscal year. During FY 00, the number of FOI requests closed was 1,146 compared to 834 in FY 99. The total number of FOI requests pending in ODE at the end of FY 00 is 621 compared to 771 in FY 99.
Congressional interest in ODE programs continued to be strong in FY 00. ODE staff responded to inquiries and participated in briefings on such topics as hearing aids, breast implants, drug test kits, dental amalgam/illness, hemodialysis, and reuse. ODE also participated in Congressional hearings held during FY 00 dealing with FDA’s budget, FDAMA, reuse, and genetic testing.
During FY 00, ODE staff authored 20 manuscripts for publication in professional and scientific journals and delivered 58 presentations at professional, scientific and trade association meetings. See Appendix B for a bibliography of publications.
In FY 00, ODE, in conjunction with the regulatory industry, sponsored one Vendor Day - an informative exhibit and exchange seminar with device manufacturers on cardiovascular, general and restorative, clinical laboratory and other devices.
In FY 00, ODE continued its Site Visit Program that was developed to enhance reviewer knowledge of how specific medical devices are designed, manufactured, and tested. The program continued to include not only visits to medical device manufacturing firms but also hospitals for the observation of certain devices in use. As a result, 11 firms and/or hospitals were visited to learn about orthopedic products, blood-glucose products, endovascular grafts, dialysis systems, IVD products, condoms, and other devices.
ODE employees attended many courses, lecturers, and grand rounds sponsored by the CDRH Staff College. Supervisors continued to participate in monthly meetings to discuss current management issues, and all employees attended all-hands meetings to learn about new FDAMA polices and procedures.
ODE sponsored three in-house training courses for employees and managers: Media Relations Workshop, Congressional Hearings Workshop and Interviewing Techniques.
ODE continues to improve and enhance its mentoring program. The program is designed to orient new employees to their job responsibilities and their workplace. The program matches new employees with a mentor who is expected to provide technical, informational and career guidance to the employee in an effort to ensure appropriate employee development. The ODE PMO Office has served as an informal mentoring agent for minorities to facilitate their assimilation into the workforce.
In FY 2000, ODE continued and expanded the ODE Intern Program. The program allows 4-5 college students to work in a practical work environment, gain entry level professional "real work" experience and work alongside some the Agency’s top healthcare authorities. Special attention is given to minority candidates. ODE continues to expand the program to include American and foreign professionals.
ODE, along with a sister organization, the Office of Health Industry Programs, continued the DSMA/ODE Exchange Program, an internal program that allows scientific reviewers from each Office to exchange places for a period of 60-90 days. Each participant is expected to learn about the operations and integral workings of the other Office.
ODE established the ODE Employee Exchange Program. The primary purpose of the program is to allow staff members the opportunity to work in other Offices and Centers within FDA to keep abreast of current advances and practices in sister organizations, as well as changes in legislation, regulations, scientific and legislative literature in other medical fields.
In an effort to increase the hiring of minorities within the Center, ODE participated in various recruitment and job fairs including the President’s Committee on the Employment of People with Disabilities Job Fair and the Hispanic Association of Colleges and Universities (HACU) Employment Fair.
ODE tracking system changes included premarket database enhancements, revised query programs and performance reports, and the development and implementation of the CLIA categorization tracking system, the 510(k) Exempt CLIA submission tracking system, and the 513(g) (device determination) tracking system. In addition, revisions were made in the Classification database for the expanded third party review and to the PMA modular review tracking system. The ODE division tracking system was updated to accommodate CLIA and 513(g) submissions and to produce new reports. All tracking systems were modified to reflect the division reorganizations of ODE.
ODE continued to upgrade equipment in order to improve the processing of applications and interactions with the regulated industry and the public. Speakerphones, Windows NT on all PCs, laptops, PCs, laser printers, uninterruptible power supplies, and facsimile machines were among the improvements. In addition, ODE contributed funds to upgrade the Center’s telephone system to enable dial-in access speeds to approach 56K and to help with the development of a new storage system for archived documents.
In FY 00, ODE received 113 electronic submissions for PMAs, IDEs, and 510(k)s from 37 different sponsors. ODE reviewers received parts of submissions in electronic format such as additional information, summaries of safety and effectiveness, and proposed labeling and those submissions were recorded as electronic submissions. Prior contact with an ODE division is requested before developing and sending an electronic submission. Instructions for submitting electronic submissions can be found on the FDA home page at the address http://www.fda.gov/cdrh/elecsub.html.
The ODE use of videoconferencing to interact with the regulated industry continued to show limited use. In FY 00, 8 videoconferences were held involving industry, other Federal agencies and professional societies. CDRH has the ability to conduct Room and Desktop Video Conferences with outside parties that have H.320 compliant systems, a standard for video conferencing over ISDN lines and other narrowband transmission media.
ODE continues to provide information on the web that can be downloaded and searched through the CDRH home page at http://www.fda.gov/cdrh. Information on Premarket Approval Applications (PMAs) and Premarket Notifications (510(k)s) can be found under the Popular Items/New Device Information on the CDRH home page.
Anyone can search the Releasable 510(k) and PMA databases, download 510(k) or PMA files, obtain the monthly PMA, HDE and 510(k) listings and Summaries of Safety and Effectiveness Data, and read about the "Real-Time" program for PMA supplements. A database of guidance documents is available at the address http://www.fda.gov/cdrh/ggpmain.html. The database is searchable by words in the document title, office, division, or any combination of these elements. In FY00, ODE posted 39 guidance documents on the web. In addition, information on ODE’s panel meeting schedules and summaries can be found on the internet at http://www.fda.gov/cdrh/panelmtg.html.
Center for Devices and Radiological Health (CDRH) maintains searchable databases of devices previously approved for marketing or declared substantially equivalent to a legally marketed device at http://www.fda.gov/cdrh/mda/mda-databases.html.
The Consumer Staff in FDA’s Center for Devices and Radiological Health, Division of Small Manufacturers Assistance also provides information to consumers regarding medical devices and radiation-emitting products to enhance their ability to avoid risk, achieve maximum benefit, and make informed decisions about the use of such products.
| Website: | http://www.fda.gov/cdrh/consumer/index.html |
| E-Mail: | dsma@cdrh.fda.gov |
| Phone: | Toll Free 1-888-463-6332 or 301-827-3990 directly between the hours of 8:00 a.m. – 4:30 p.m. EST |
The FDA Breast Implant website for consumer information is available at http://www.fda.gov/cdrh/breastimplants/index.html.
A new CDRH website entitled LASIK Eye Surgery: Learning About LASIK is available at http://www.fda.gov/cdrh/lasik/.
[NOTE: Although accurate at the time of publication, the data in the following tables may change slightly in subsequent reports to reflect changes in the regulatory status of submissions or verification of data entry. There are also likely to be changes in the previous years’ annual report numbers in tables representing receipt cohort data. For example, if an incoming PMA supplement is later converted to an original PMA, changes are made in the appropriate tables. Likewise, some data from earlier reporting periods may have been changed to reflect similar corrections in data entry. These adjustments are not likely to have a significant effect on conclusions based on these data. Percentages of actions are presented in some tables. They may not add up to 100% in all cases due to the rounding off of fractions.] Refer to Tables 1 (page 14) and 2 (page 15) for general summary of major submissions received and completed.
Table 3.
PMA/HDE/IDE/510(k) Submissions Received
FY 96 - FY 00
| Type of Submission | Number Received | ||||
FY 96 |
FY 97 |
FY 98 |
FY 99 |
FY 00 |
|
Premarket Approval (PMAs) |
|||||
| Original Applications | 44 | 66 | 47 | 60 | 67 |
| Amendments | 883 | 829 | 710 | 767 | 978 |
| Supplements | 415 | 409 | 513 | 552 | 545 |
| Amendments to Supplements | 823 | 819 | 863 | 924 | 932 |
| Reports for Orig. Applications | 435 | 435 | 431 | 406 | 419 |
| Reports for Supplements | 24 | 2 | 0 | 0 | 0 |
| Master Files | 65 | 130 | 94 | 25 | 44 |
| PMA Subtotal | 2,689 | 2,690 | 2,658 | 2,734 | 2,985 |
| Humanitarian Device Exemptions (HDEs) | |||||
| Original Applications | 0 | 4 | 8 | 12 | 11 |
| Amendments | 0 | 10 | 32 | 55 | 56 |
| Supplements | 0 | 0 | 0 | 4 | 10 |
| Amendments to Supplements | 0 | 0 | 0 | 3 | 12 |
| Reports for Orig. Applications | 0 | 0 | 0 | 6 | 9 |
| Reports for Supplements | 0 | 0 | 0 | 0 | 0 |
| HDE Subtotal | 0 | 14 | 40 | 80 | 98 |
| Investigational Device Exemptions (IDEs) | |||||
| Original Applications | 253 | 297 | 322 | 304 | 311 |
| Amendments | 219 | 223 | 226 | 275 | 240 |
| Supplements | 3,189 | 3,776 | 4,277 | 4,127 | 4,388 |
| IDE Subtotal | 3,661 | 4,296 | 4,825 | 4,706 | 4,939 |
| Premarket Notification (510(k)s) | |||||
| Original Notifications | 5,297 | 5,049 | 4,623 | 4,458 | 4,202 |
| Supplements | 3,246 | 2,785 | 2,023 | 1,872 | 1,742 |
| Amendments | 5,343 | 4,433 | 3,692 | 2,962 | 2,953 |
| 510(k) Subtotal | 13,886 | 12,267 | 10,338 | 9,292 | 8,897 |
| PMA/HDE/IDE/510(k) Total | 20,236 | 19,267 | 17,861 | 16,812 | 16,919 |
Table 4. Original
PMA Decision Cohort Performance*
FY 96 - FY 00
FY 96 |
FY 97 |
FY 98 |
FY 99 |
FY 00 |
|
Number Received |
44 |
0 |
55 |
72 |
67 |
| PMA Actions Filing Decisions | |||||
| Filed | 45 | 58 | 51 | 65 | 64 |
| Not Filed | 17 | 16 | 10 | 7 | 4 |
| Others | 0 | 0 | 0 | 0 | 0 |
| Filing Decision Subtotal | 62 | 74 | 61 | 72 | 68 |
| Scientific Review Decisions | |||||
| Major Deficiencies | 32 | 38 | 28 | 32 | 51 |
| Minor Deficiencies | 5 | 5 | 10 | 4 | 11 |
| Othera | 97 | 138 | 105 | 105 | 111 |
| Scientific Review Decisions Subtotal | 134 | 181 | 143 | 141 | 173 |
| Approval Decisions | |||||
| Approvals | 43 | 48 | 46 | 45 | 43 |
| Approvable | 27 | 14 | 7 | 7 | 33 |
| Not Approvable | 6 | 5 | 12 | 1 | 4 |
| Denials | 0 | 0 | 0 | 0 | 0 |
| Approval Decision Subtotal | 76 | 67 | 65 | 53 | 80 |
| Total PMA Actions | 272 | 322 | 269 | 266 | 321 |
| Average Review Time (Days) | |||||
| for Approvalsb | |||||
| FDA | 289 | 207 | 154 | 149 | 158 |
| Non-FDA | 55 | 40 | 37 | 26 | 40 |
| Total | 343 | 247 | 191 | 175 | 198 |
| Average Elapsed Time (Days) | |||||
| for Approvalsc | |||||
| FDA | 572 | 375 | 265 | 280 | 244 |
| Non-FDA | 214 | 122 | 108 | 100 | 119 |
| Total | 786 | 497 | 373 | 380 | 363 |
| Number under Review at End of Periodd | |||||
| Activee | 57 | 44 | 29 | 49 | 35 |
| (Active and overdue) | (17) | (0) | (0) | (0) | (0) |
| On holdf | 39 | 41 | 41 | 38 | 41 |
| Total | 96 | 85 | 70 | 87 | 76 |
| _____________________ | |
| */ | For FY 97, 98 and 99, PMA data includes a special category of PMAs. Humanitarian Devices Exemption (HDE) applications are similar in both form and content to PMAs but are exempt from the effectiveness requirements of PMAs. An approved HDE authorizes marketing of the humanitarian use device. |
| a/ | Includes actions that did not result in an approval/denial decision, such as GMP deficiency letters prior to inspection, an applicant directed hold, reclassification of the device and conversion of the PMA to another regulatory category, or official correspondence concerning the abandonment or withdrawal of the PMA, placing the PMA on hold, and other miscellaneous administrative actions. |
| b/ | Average review times are calculated under the Premarket Approval of Medical Devices Regulation (21 CFR Part 814). Under this regulation, the review clock is reset upon FDA’s receipt of a "major amendment" or a response to a "refuse to file" letter. Thus, average review time, unlike average elapsed time, excludes all review times that occurred prior to the latest resetting of the clock. Number of months based upon 30.4 day/month and rounded to one decimal point. |
| c/ | The average elapsed time includes all increments of time a PMA was under review, including all of the increments of time it was under review by FDA and all increments of time it was on hold, during which time it was being worked on by the manufacturer. Thus the average elapsed time is the average time taken to obtain approval of a PMA from its filing date until it receives final approval. |
| d/ | The number under review at the end of a period may not reconcile with the number under review at the end of the previous period (plus receipts less approvals) because of deletions and conversions not reflected in the table. |
| e/ | FDA responsible for processing application. |
| f/ | FDA processing of applications officially suspended pending receipt of additional information from the applicant. |
Table 5. Original
PMA Receipt Cohort Performance*
FY 96 – FY 00
FY 96 |
FY 97 |
FY 98 |
FY 99 |
FY 00 |
||||||
Original PMAs Received |
||||||||||
| PMAs | 37 |
46 | 32 | 48 | 42 | |||||
| Expedited PMAs | 5 | 10 | 6 | 7 | 4 | |||||
| Total | 42 | 56 | 38 | 55 | 46 | |||||
| Filing Decisionsa | ||||||||||
| Filed | 42 | 56 | 38 | 55 | 46 | |||||
| Not Filed | 11 | 8 | 3 | 1 | 2 | |||||
| Number (%) of Filing/Not Filing | ||||||||||
| Decisions within 45 Days | 26 | (49) | 51 | (80) | 30 | (73) | 44 | (79) | 39 | (81) |
| Average Days/Cycle | 67 | 39 | 44 | 42 | 40 | |||||
| Final Actionsb | ||||||||||
| Approvals | 28 | 45 | 26 | 38 | 12 | |||||
| Denials | 0 | 0 | 0 | 0 | 0 | |||||
| Otherc | 18 | 22 | 15 | 9 | 8 | |||||
| Total | 46 | 67 | 41 | 47 | 20 | |||||
| Filing to First Action Excluding withdrawals, conversions, etc.d | ||||||||||
| Number Received and Filed | 42 | 56 | 38 | 55 | 46 | |||||
| Number of First Actions | 37 | 53 | 37 | 55 | 41 | |||||
| Average FDA Days | 195 | 147 | 134 | 145 | 139 | |||||
| Median FDA Days | 187 | 175 | 145 | 147 | 160 | |||||
| Number (%) of First Actions within 180 Days |
18 |
(43) |
41 |
(73) |
32 |
(84) |
43 |
(78) |
41 |
(89) |
| Filing to First Action Including withdrawals, conversions, etc.e | ||||||||||
| Number Received and Filed | 42 | 56 | 38 | 55 | 46 | |||||
| Number of First Actions | 42 | 56 | 38 | 55 | 46 | |||||
| Average FDA Days | 228 | 146 | 134 | 145 | 140 | |||||
| Median FDA Days | 183 | 173 | 141 | 147 | 152 | |||||
| Number (%) of First Actions within 180 Days |
20 |
(48) |
43 |
(77) |
33 |
(87) |
43 |
(78) |
46 |
(100) |
| Filing to Final Actions Excluding withdrawals, conversions, etc.f | ||||||||||
| Number Received and Filed | 42 | 56 | 38 | 55 | 46 | |||||
| Number of Final Actions | 30 | 45 | 28 | 35 | 15 | |||||
| Average FDA (Total) Elapsed Time |
358 |
(514) |
264 |
(346) |
239 |
(321) |
244 |
(293) |
164 |
(214) |
| Median FDA (Total) Elapsed Time |
329 |
(408) |
217 |
(287) |
198 |
(201) |
240 |
(269) |
178 |
(211) |
| Number (%) of Final Actions within 180 FDA Days |
6 |
(20) |
19 |
(42) |
12 |
(43) |
7 |
(20) |
11 |
(73) |
| Number (%) of Final Actions within 180 Total Days |
4 |
(13) |
16 |
(36) |
10 |
(36) |
5 |
(14) |
5 |
(33) |
| Filing to Final Action Including withdrawals, conversions, etc.g | ||||||||||
| Number Received and Filed | 42 | 56 | 38 | 55 | 46 | |||||
| Number of Final Actions | 42 | 54 | 34 | 38 | 23 | |||||
| Average FDA (Total) Elapsed Time |
378 |
(530) |
257 |
(373) |
224 |
(355) |
245 |
(303) |
161 |
(200) |
| Median FDA (Total) Elapsed Time | 321 | (420) | 200 | (315) | 187 | (252) | 243 | (271) | 175 | (195) |
| Number (%) of Final Actions within 180 FDA Days |
8 |
(19) |
23 |
(43) |
17 |
(50) |
8 |
(21) |
18 |
(78) |
| Number (%) of Final Actions within 180 Total Days |
6 |
(14) |
18 |
(33) |
11 |
(32) |
5 |
(13) |
10 |
(44) |
| Average Number of FDA Cycles from Receipt to Final Action Including |
||||||||||
| Withdrawals, conversions, etc b. |
|
|
|
|
|
|||||
| Percentile FDA Days from Filing to First Actione | ||||||||||
| 25th | 165 | 111 | 99 | 115 | 113 | |||||
| 50th (Median) | 183 | 173 | 141 | 147 | 152 | |||||
| 75th | 231 | 180 | 174 | 179 | 176 | |||||
| 90th | 316 | 199 | 181 | 227 | 179 | |||||
| Percentile FDA Days from Filing to First Actiond | ||||||||||
| 25th | 171 | 118 | 99 | 115 | 116 | |||||
| 50th (Median) | 187 | 175 | 145 | 147 | 160 | |||||
| 75th | 252 | 182 | 175 | 179 | 179 | |||||
| 90th | -- | 217 | 192 | 227 | -- | |||||
| Percentile FDA (Total) Days from Filing to Final Actiong | ||||||||||
| 25th | 233 | (272) | 165 | (177) | 141 | (158) | 185 | (236) | 120 | (175) |
| 50th (Median) | 321 | (420) | 200 | (315) | 187 | (252) | 243 | (271) | 175 | (195) |
| 75th | 432 | (785) | 382 | (520) | 289 | (564) | 284 | (384) | 181 | (234) |
| 90th | 712 | (961) | 440 | (708) | 392 | (789) | 341 | (481) | 202 | (280) |
| Percentile FDA (Total) Days from Filing to Final Actionf | ||||||||||
| 25th | 233 | (272) | 170 | (177) | 157 | (158) | 185 | (234) | 147 | (178) |
| 50th (Median) | 329 | (408) | 217 | (287) | 198 | (201) | 240 | (269) | 178 | (211) |
| 75th | 419 | (779) | 390 | (520) | 328 | (387) | 284 | (372) | 181 | (240) |
| 90th | 710 | (987) | 440 | (680) | 392 | (801) | 341 | (437) | 228 | (280) |
| Number pending as of 9/30/00 | ||||||||||
| Active | 0 | 1 | 0 | 8 | 8 | |||||
| Active and Overdue | 0 | 0 | 0 | 0 | 0 | |||||
| On holdh | 0 | 1 | 7 | 9 | 16 | |||||
| Total | 0 | 2 | 7 | 17 | 24 | |||||
| Summary of PMA Receipt Cohort | ||||||||||
| Approved | 28 | 45 | 26 | 38 | 12 | |||||
| Denied | 0 | 0 | 0 | 0 | 0 | |||||
| Withdrawn | 11 | 11 | 9 | 3 | 6 | |||||
| Other | 7 | 11 | 6 | 6 | 2 | |||||
| Under Review | 0 | 1 | 0 | 8 | 8 | |||||
| On Holdh | 0 | 1 | 7 | 9 | 16 | |||||
| Total | 46 | 69 | 48 | 64 | 44 | |||||
| _____________________ | |
| */ | For each fiscal year, September 30, 2000 was used as the cutoff date. The FY 00 cohort represents only receipts through March 31, 2000 (first six months of the fiscal year). The average elapsed time includes all increments of time a PMA was under review, including all of the increments of time it was under review by FDA and all increments of time it was on hold, during which time it was being worked on by the manufacturer. Thus the average elapsed time is the average time taken to obtain approval of a PMA from its filing date until it receives final approval. |
| a/ | The filing decision represents the count of applications with a filing date within the fiscal year as of the cutoff date. For example, a PMA that is considered complete at the time of submission would have a received date equal to the filed date. However, if the agency refuses to file the PMA, it is considered incomplete and the filed date becomes the date of the amendment that makes the submission complete for filing. Therefore, it is possible that the submission may be received in one fiscal year but not be considered a filed PMA until a subsequent fiscal year. For the purpose of receipt cohort reporting, PMAs are considered "received" based on the filing date rather than the receipt date. |
| b/ | The final action analyses include actions as of the cutoff date for PMAs received within the fiscal year. |
| c/ | Includes only actions that resulted in withdrawal, conversion, and other final actions not resulting in approval or denial. |
| d/ | The first action analyses include actions as of the cutoff date for PMAs that were filed within the fiscal year. This measure excludes PMAs with a final action of withdrawal, conversion, or other final actions. |
| e/ | The first action analyses include actions as of the cutoff date for PMAs that were filed within the fiscal year. This measure includes PMAs with any final action including approval, denial, withdrawal, conversion, or other final actions. |
| f/ | The final actions analyses include actions as of the cutoff date for PMAs that were filed within the fiscal year. This measure excludes PMAs with a final action of withdrawal, conversion, or other final action not resulting in approval or denial. |
| g/ | The final actions analyses include actions as of the cutoff date for PMAs that were filed within the fiscal year. This measure includes PMAs with any final action including approval, denial, withdrawal, conversion, or other final actions. |
| h/ | "On hold" describes the FDA processing of applications officially suspended pending receipt of additional information from the applicant. |
Table 6. PMA
Supplement Decision Cohort Performance*
FY 96 - FY 00
FY 96 |
FY 97 |
FY 98 |
FY 99 |
FY 00 |
|
Number Received |
415 |
409 |
513 |
556 |
545 |
| PMA Supplement Actions Panel Track Filing Decisionsa |
|||||
| Filed | 8 | 15 | 7 | 17 | 14 |
| Not Filed | 1 | 1 | 2 | 2 | 3 |
| Other | 0 | 0 | 0 | 0 | 0 |
| Filing Decision Subtotal | 9 | 16 | 9 | 19 | 17 |
| Scientific Review Decisions | |||||
| Major Deficiencies | 9 | 3 | 4 | 12 | 14 |
| Minor Deficiencies | 1 | 1 | 2 | 0 | 1 |
| Otherb | 141 | 128 | 62 | 60 | 83 |
| Scientific Review Decisions Subtotal | 151 | 132 | 68 | 72 | 98 |
| Approval Decisions | |||||
| Panel track approvalsc | 0 | 4 | 5 | 11 | 12 |
| Nonpanel track approvals | 462 | 397 | 416 | 426 | 462 |
| Approvable | 33 | 49 | 47 | 25 | 100 |
| Not approvable | 48 | 76 | 63 | 62 | 58 |
| Approval Decision Subtotal | 543 | 526 | 531 | 524 | 632 |
| Total PMA Supplement Actions | 703 | 674 | 608 | 615 | 747 |
| Average Review Time (Days) for Approvalsd | |||||
| FDA | 146 | 100 | 82 | 76 | 76 |
| Non-FDA | 36 | 12 | 25 | 16 | 18 |
| Total | 182 | 112 | 107 | 92 | 94 |
| Average Elapsed Time (Days) for Approvalse | |||||
| FDA | 167 | 120 | 109 | 92 | 95 |
| Non-FDA | 49 | 23 | 43 | 26 | 27 |
| Total | 216 | 143 | 153 | 118 | 122 |
| Number under Review at End of Periodf | |||||
| Activeg | 162 | 110 | 139 | 158 | 98 |
| (Active and overdue) | (17) | (0) | (0) | (0) | (0) |
| On holdh | 74 | 80 | 57 | 70 | 84 |
| Total | 236 | 190 | 196 | 228 | 182 |
| _________________ | |
| */ | For FY 97, 98 and 99, PMA data includes a special category of PMAs. Humanitarian Devices Exemption (HDE) applications are similar in both form and content to PMAs but are exempt from the effectiveness requirements of PMAs. An approved HDE authorizes marketing of the humanitarian use device. |
| a/ | Filing and not filing decisions are for panel track PMA supplements only. Nonpanel track PMA supplements are automatically filed upon receipt. |
| b/ | Includes actions that did not result in an approval/denial decision, such as GMP letters prior to inspection, an applicant directed hold, reclassification of the device and conversion of the PMA supplement to another regulatory category, and official correspondence concerning the abandonment or withdrawal of the supplement, the status of the supplement as a special (changes being effected) or 30-day submission, and other miscellaneous administrative actions. |
| c/ | Panel track supplements are subject to the full administrative procedures normally associated with original PMAs, i.e., panel review, preparation of a summary of safety and effectiveness. |
| d/ | Average review times are calculated under the Premarket Approval of Medical Devices Regulation (21 CFR Part 814). Under this regulation, the review clock is reset upon FDA’s receipt of a "major amendment" or a response to a "refuse to file" letter. Thus, average review time, unlike average elapsed time, excludes all review times that occurred prior to the latest resetting of the clock. Number of months based upon 30.4 day/month and rounded to one decimal point. |
| e/ | The average elapsed time includes all increments of time a PMA was under review, including all of the increments of time it was under review by FDA and all increments of time it was on hold, during which time it was being worked on by the manufacturer. Thus the average elapsed time is the average time taken to obtain approval of a PMA from its filing date until it receives final approval. |
| f/ | The number under review at the end of a period may not reconcile with the number under review at the end of the previous period (plus receipts less approvals) because of deletions and conversions which are not reflected in the table. |
| g/ | FDA responsible for processing application. |
| h/ | FDA’s processing of application officially suspended pending receipt of additional information from the applicant. |
Table 7. PMA
Supplement Receipt Cohort Performance*
FY 96 - FY 00
FY 96 |
FY 97 |
FY 98 |
FY 99 |
FY 00 |
||||||
PMA Supplements Received |
||||||||||
| PMA Supplements | 410 | 402 | 510 | 541 | 270 | |||||
| Expedited PMA Supplements | 3 | 3 | 1 | 6 | 1 | |||||
| Total | 413 | 405 | 511 | 547 | 271 | |||||
| Filing Decisionsa | ||||||||||
| Filed | 4 | 6 | 9 | 15 | 7 | |||||
| Not Filed | 0 | 1 | 1 | 0 | 1 | |||||
| Number of Filing/Not Filing | ||||||||||
| Decisions within 45 Days | 3 | 5 | 9 | 10 | 7 | |||||
| Average Days/Cycle | 45 | 45 | 42 | 45 | 43 | |||||
| PMA Supplement Final Actionsb | ||||||||||
| Approvals | 379 | 369 | 427 | 441 | 200 | |||||
| Denials | 0 | 0 | 0 | 0 | 0 | |||||
| Otherc | 34 | 36 | 82 | 84 | 47 | |||||
| Filing to First Action Excluding withdrawals, conversions, etc.d | ||||||||||
| Number Received and Filed | 413 | 405 | 511 | 547 | 271 | |||||
| Number of First Actions | 398 | 396 | 491 | 526 | 265 | |||||
| Average | 122 | 90 | 82 | 74 | 67 | |||||
| Median | 130 | 73 | 59 | 39 | 42 | |||||
| Number (%) of First Actions within 180 Days |
311 |
(75) |
350 |
(86) |
433 |
(87) |
475 |
(87) |
262 |
(97) |
| Filing to First Action Including withdrawals, conversions, etc.e | ||||||||||
| Number Received and Filed | 413 | 405 | 511 | 547 | 271 | |||||
| Number of First Actions | 411 | 405 | 509 | 544 | 271 | |||||
| Average | 121 | 91 | 80 | 74 | 67 | |||||
| Median | 126 | 70 | 49 | 37 | 39 | |||||
| Number (%) of First Actions within 180 Days |
322 | (78) | 357 | (88) | 460 | (90) | 49 | 1(90) | 268 | (99) |
| Average Number of FDA Cycles from Receipt to Final Action Including |
||||||||||
| withdrawals, conversions, etc.b | 1.2 | 1.1 | 1.1 | 1.1 | 1.0 | |||||
| Filing to Final Action Excluding withdrawals, conversions, etc.f | ||||||||||
| Number Received and Filed | 413 | 405 | 511 | 547 | 271 | |||||
| Number of Final Actions | 379 | 367 | 460 | 486 | 240 | |||||
| Average FDA (Total) Review Days |
148 | (185) | 106 | (129) | 94 | (118) | 79 | (95) | 66 | (76) |
| Median FDA (Total) Review Days |
132 | (150) | 70 | (83) | 49 | (66) | 35 | (47) | 35 | (43) |
| Number (%) of Final Actions Within 180 Days |
259 |
(68) |
304 |
(83) |
374 |
(81) |
420 |
(86) |
234 |
(98) |
| Number (%) of Final Actions Within 180 Total Days |
236 |
(62) |
286 |
(78) |
352 |
(77) |
406 |
(84) |
225 |
(94) |
| Filing to Final Action Including withdrawals, conversions, etc.g | ||||||||||
| Number Received and Filed | 413 | 405 | 511 | 547 | 271 | |||||
| Number of Final Actions | 413 | 400 | 503 | 517 | 247 | |||||
| Average FDA (Total) Review Days |
147 | (202) | 110 | (147) | 97 | (131) | 82 | (102) | 66 | (75) |
| Median FDA (Total) Review Days |
132 | (156) | 74 | (94) | 51 | (69) | 36 | (50) | 35 | (43) |
| Number (%) of Final Actions Within 180 Days |
284 |
(69) |
324 |
(81) |
490 |
(97) |
447 |
(87) |
241 |
(98) |
| Number (%) of Final Actions Within 180 Total Days |
249 |
(60) |
296 |
(74) |
371 |
(74) |
424 |
(82) |
232 |
(94) |
| Percentile FDA Days from Filing to First Actione | ||||||||||
| 25th | 57 | 29 | 22 | 19 | 20 | |||||
| 50th (Median) | 126 | 70 | 49 | 37 | 39 | |||||
| 75th | 179 | 155 | 156 | 140 | 113 | |||||
| 90th | 196 | 181 | 180 | 181 | 165 | |||||
| Percentile FDA Days from Filing to First Actiond | ||||||||||
| 25th | 63 | 32 | 22 | 20 | 21 | |||||
| 50th (Median) | 130 | 73 | 59 | 39 | 42 | |||||
| 75th | 180 | 165 | 169 | 151 | 116 | |||||
| 90th | 201 | 182 | 183 | 190 | 190 | |||||
| Percentile FDA (Total) Days from Filing to Final Actiong | ||||||||||
| 25th | 63 | (76) | 33 | (36) | 22 | (24) | 20 | (24) | 18 | (24) |
| 50th (Median) | 132 | (156) | 74 | (94) | 51 | (69) | 36 | (50) | 35 | (43) |
| 75th | 187 | (225) | 169 | (182) | 175 | (182) | 147 | (161) | 109 | (118) |
| 90th | 296 | (446) | 216 | (351) | 212 | (322) | 190 | (232) | 165 | (172) |
| Percentile FDA (Total) Days from Filing to Final Actionf | ||||||||||
| 25th | 64 | (76) | 32 | (35) | 22 | (25) | 19 | (24) | 18 | (24) |
| 50th (Median) | 132 | (150) | 70 | (83) | 49 | (66) | 35 | (47) | 35 | (43) |
| 75th | 187 | (210) | 162 | (179) | 175 | (179) | 141 | (152) | 108 | (117) |
| 90th | 303 | (379) | 207 | (313) | 213 | (281) | 190 | (214) | 165 | (174) |
| Number under review as of 9/30/00 | ||||||||||
| Active | 0 | 0 | 0 | 5 | 4 | |||||
| Active and Overdue | 0 | 0 | 0 | 2 | 0 | |||||
| On holdh | 0 | 5 | 8 | 25 | 20 | |||||
| Total | 0 | 5 | 8 | 32 | 24 | |||||
| Summary of PMA Supplement Receipt Cohort | ||||||||||
| Approved | 379 | 369 | 427 | 441 | 200 | |||||
| Denied | 0 | 0 | 0 | 0 | 0 | |||||
| Withdrawn | 28 | 28 | 30 | 32 | 6 | |||||
| Other | 6 | 8 | 52 | 52 | 41 | |||||
| Under Review | 0 | 0 | 0 | 7 | 4 | |||||
| On Holdh | 0 | 5 | 8 | 25 | 20 | |||||
| Total | 413 | 410 | 517 | 557 | 271 | |||||
| _____________________ | |
| */ | For each fiscal year, September 30, 2000 was used as the cutoff date. The FY 00 cohort represents only receipts through March 31, 2000 (first six months of the fiscal year). The average elapsed time includes all increments of time a PMA was under review, including all of the increments of time it was under review by FDA and all increments of time it as on hold, during which time it was being worked on by the manufacturer. Thus the average elapsed time is the average time taken to obtain approval of a PMA from its filing date until it receives final approval. |
| a/ | Filing and not filing decisions are for panel track PMA supplements only. Nonpanel track PMA supplements are automatically filed upon receipt |
| b/ | The final action analyses include actions as of the cutoff date for PMA supplements received within the fiscal year. |
| c/ | Includes only actions that resulted in withdrawal, conversion, and other final actions not resulting in approval or denial. |
| d/ | The first action analyses include actions as of the cutoff date for PMA supplements that were filed within the fiscal year. This measure excludes PMA supplements with a final action of withdrawal, conversion, or other final actions. |
| e/ | The first action analyses include actions as of the cutoff date for PMA supplements that were filed within the fiscal year. This measure includes PMA supplements with any final action including approval, denial, withdrawal, conversion, or other final actions. |
| f/ | The final actions analyses include actions as of the cutoff date for PMA supplements that were filed within the fiscal year. This measure excludes PMA supplements with a final action of withdrawal, conversion, or other final action not resulting in approval or denial. |
| g/ | The final actions analyses include actions as of the cutoff date for PMA supplements that were filed within the fiscal year. This measure includes PMA supplements with any final action including approval, denial, withdrawal, conversion, or other final actions. |
| h/ | "On hold" describes the FDA processing of applications officially suspended pending receipt of additional information from the applicant. |
Table 8. HDE
Submissions Received
FY97 – FY00
| Type of Submission | Number Received |
|||
FY 97 |
FY 98 |
FY 99 |
FY 00 |
|
Humanitarian Device |
||||
| Exemption (HDE) | ||||
| Original Applications | 4 |
8 |
12 |
11 |
| Amendments | 10 |
32 |
55 |
56 |
| Supplements | 0 |
0 |
4 |
10 |
| Amendments to Supplements | 0 |
0 |
3 |
12 |
| Reports for Orig. Applications | 0 |
0 |
6 |
9 |
| Reports for Supplements | 0 |
0 |
0 |
0 |
| HDE Subtotal | 14 |
40 |
80 |
98 |
Table 9. Original
HDE Decision Cohort Performance
FY97 – FY00
FY 97 |
FY 98 |
FY 99 |
FY 00 |
|
Number Received |
4 |
8 |
12 |
11 |
| HDE Actions | ||||
| Filing Decisions | ||||
| Filed | 2 | 9 | 10 | 8 |
| Not Filed | 0 | 1 | 1 | 4 |
| Othera | 0 | 1 | 1 | 0 |
| Filing Decision Subtotal | 2 | 11 | 12 | 12 |
| Scientific Review Decisions | ||||
| Major Deficiencies | 0 | 0 | 6 | 7 |
| Minor Deficiencies | 1 | 1 | 0 | 3 |
| Otherb | 0 | 0 | 4 | 6 |
| Scientific Review Decisions Subtotal | 1 | 1 | 10 | 16 |
| Approval Decisions | ||||
| Approvals | 2 | 4 | 6 | 6 |
| Approvable | 0 | 0 | 5 | 1 |
| Not Approvable | 0 | 0 | 0 | 0 |
| Denials | 0 | 0 | 0 | 0 |
| Approval Decision Subtotal | 2 | 4 | 11 | 7 |
| Other Final Decisionsc | 0 | 2 | 4 | 1 |
| Total HDE Actions | 5 | 18 | 37 | 36 |
| Filing to First Actiond | ||||
| Number of First Actions | 2 | 6 | 13 | 8 |
| Average Number of FDA Days | 68 |
139 |
87 |
61 |
| Number of First Actions Within 75 Days |
1 |
1 |
7 |
8 |
| Average Elapsed Time (Days) for Approvalse | ||||
| FDA | 108 |
152 |
113 |
112 |
| Non-FDA | 12 |
0 |
50 |
104 |
| Total | 120 |
152 |
163 |
216 |
| |
||||
| Average Number of FDA Cycles from Receipt to Final Actionf | 1 |
1.2 |
1.2 |
1.3 |
Number under Review at End of Period |
||||
| Active | 2 |
3 |
2 |
2 |
| Active and overdue | 0 |
0 |
0 |
0 |
| On hold | 0 |
1 |
8 |
8 |
| Total | 2 |
4 |
10 |
10 |
| _________________ | |
| a/ | Includes final actions, such as withdrawal or conversion to another regulatory category, that occur prior to a filing decision being made. |
| b/ | Includes actions that did not result in a final decision, such as GMP deficiency letter or an applicant-directed hold. |
| c/ | Includes final actions other than approval or denial, such as withdrawal or conversion to another regulatory category. |
| d/ | First actions may include major and minor deficiency decisions; approvable, not approvable, approval and denial decisions; receipt of an unsolicited major amendment; and other final actions, such as withdrawal or conversion to another regulatory category. |
| e/ | The average amount of time taken to obtain approval of an HDE from the filing date until final approval. |
| f/ | A cycle is counted as the initial submission and each resetting of FDA’s review clock, such as a response to a non-filing decision or the submission of a major amendment. |
| g/ | The number under review at the end of a period may not reconcile with the number under review at the end of the previous period (plus receipts less approvals) because of deletions and conversions not reflected in the table. |
| h/ | The application is under review by FDA. |
| i/ | FDA’s review of the application is officially suspended pending receipt of additional information from the applicant. |
Table 10. HDE
Supplement Decision Cohort Performance
FY97 – FY00
FY 97 |
FY 98 |
FY 99 |
FY 00 |
|
Number Received |
|
|
|
|
| HDE Supplement Actions | ||||
| Scientific Review Decisions | ||||
| Major Deficiencies | 0 |
0 |
1 |
0 |
| Minor Deficiencies | 0 |
0 |
0 |
0 |
| Othera | 0 |
0 |
2 |
0 |
| Scientific Review Decisions Subtotal | 0 |
0 |
3 |
0 |
| Approval Decisions | ||||
| Approvals | 0 |
0 |
3 |
10 |
| Approvable | 0 |
0 |
1 |
0 |
| Not Approvable | 0 |
0 |
0 |
1 |
| Denials | 0 |
0 |
0 |
0 |
| Approval Decision Subtotal | 0 |
0 |
4 |
11 |
Other Final Decisionsb |
0 |
0 |
0 |
0 |
| Total HDE Actions | 0 |
0 |
7 |
11 |
| Filing to First Actionc | ||||
| Number of First Actions | 0 |
0 |
4 |
10 |
| Average Number of FDA Days | 0 |
0 |
57 |
44 |
| Number of First Actions Within 75 Days | 0 |
0 |
4 |
10 |
| Average Elapsed Time (Days) for Approvalsd | ||||
| FDA | 0 |
0 |
70 |
43 |
| Non-FDA | 0 |
0 |
24 |
33 |
| Total | 0 |
0 |
94 |
76 |
Average Number of FDA Cycles from Receipt to Final Actione |
|
|
|
|
| Number under Review at End of Periodf | ||||
| Activeg | 0 |
0 |
0 |
0 |
| Active and overdue | 0 |
0 |
0 |
0 |
| On holdh | 0 |
0 |
1 |
1 |
| Total | 0 |
0 |
1 |
1 |
| _________________ | |
| a/ | Includes actions that did not result in a final decision, such as GMP deficiency letter or an applicant-directed hold. |
| b/ | Includes final actions other than approval or denial, such as withdrawal or conversion to another regulatory category. |
| c/ | First actions may include major and minor deficiency decisions; approvable, not approvable, approval and denial decisions; receipt of an unsolicited major amendment; and other final actions, such as withdrawal or conversion to another regulatory category. |
| d/ | The average amount of time taken to obtain approval of an HDE Supplement from the filing date until final approval. |
| e/ | A cycle is counted as the initial submission and each resetting of FDA’s review clock, such as a response to a non-filing decision or the submission of a major amendment. |
| f/ | The number under review at the end of a period may not reconcile with the number under review at the end of the previous period (plus receipts less approvals) because of deletions and conversions not reflected in the table. |
| g/ | The application is under review by FDA. |
| h/ | FDA’s review of the application is officially suspended pending receipt of additional information from the applicant. |
Table 11. Original
IDEs
FY 96 - FY 00
FY 96 |
FY 97 |
FY 98 | FY 99 |
FY 00 |
|
Number Received |
253 |
297 |
322 |
304 |
311 |
| Number of Decisions | |||||
| Approved | 171 | 172 | 201 | 176 | 213 |
| Not approved | 63 | 79 | 82 | 82 | 66 |
| Othera | 26 | 21 | 42 | 47 | 41 |
| Total | 260 | 272 | 325 | 305 | 320 |
| Percent (%) of Approvals Made during First Review Cycleb | 73 | 69 | 71 | 68 | 76 |
| Average FDA Review Time (days) | 28 | 29 | 27 | 27 | 28 |
| Percent (%) of Decisions Made within 30 Days |
99d |
100 |
100 |
99 |
99 |
| Number under Review at End of Periodc | 8 | 32 | 29 | 28 | 19 |
| Number Overdue at End of Period | 0 | 0 | 0 | 0 | 0 |
| ____________________ | |
| a/ | Includes deletions, withdrawals, and other administrative actions not resulting in an approval/disapproval decision. |
| b/ | Based on "approved" and "not approved" decisions only. |
| c/ | The number under review at the end of a period may not reconcile with the number under review at the end of the previous period (plus receipts less approvals) because of deletions and conversions which are not reflected in the table. |
| d/ | In October 1995, ODE moved its offices from Piccard Drive to Corporate Boulevard in Rockville, Maryland. ODE accepted premarketing submissions during the 14-day moving period but added 2 weeks to the due dates of IDEs. This 2-week delay is reflected in the percent of decisions made within the 30 days for original IDEs and amendments. This policy was announced in two notices in the Federal Register of October 14, 1994 (pg. 52170) and November 29, 1994 (pg. 60092). |
Table 12. IDE
Amendments
FY 96 - FY 00
FY 96 |
FY 97 |
FY 98 |
FY 99 |
FY 00 |
|
Amendments Receiveda |
219 |
223 |
226 |
275 |
240 |
| Decisions on Amendments | |||||
| Approved | 98 | 101 | 94 | 97 | 107 |
| Not approved | 29 | 25 | 36 | 42 | 34 |
| Otherb | 91 | 94 | 95 | 129 | 110 |
| Total | 218 | 220 | 225 | 268 | 251 |
| Average FDA Review Time (days) | 18 | 18 | 19 | 18 | 19 |
| Percent (%) of Decisions Made within 30 Days | 98e | 100 | 100 | 100 | 100 |
| Average Approval Time (days) for IDEs with Amendments | |||||
| FDA time | 53 | 61 | 55 | 57 | 70 |
| Non-FDA time | 78 | 84 | 35 | 88 | 66 |
| Total timec | 131 | 145 | 90 | 145 | 136 |
| Number of Amendments per Approved IDE | 1.4 | 1.8 | 1.4 | 1.6 | 2.3 |
| Amendments under Review at End of Periodd | 9 | 12 | 13 | 19 | 9 |
| Amendments Overdue at End of Period | 0 | 0 | 0 | 0 | 0 |
| ______________________ | |
| a/ | Submissions received after the original IDE and prior to approval of the IDE application. |
| b/ | Includes actions that did not result in an approval/disapproval decision, such as withdrawal of the IDE or the amendment by the sponsor, and other administrative actions, e.g., acknowledgement letters concerning the submission of information that did not require independent approval/disapproval and other administrative information, such as a change of address. |
| c/ | The average IDE approval time represents the total time it has taken, on average, for an original IDE that was initially disapproved to be approved after the submission of amendments to correct deficiencies. The time being measured here covers the period from the date the original IDE was received to the date of final approval of an IDE amendment. |
| d/ | The number under review at the end of a period may not reconcile with the number under review at the end of the previous period (plus receipts less approvals) because of deletions and conversions which are not reflected in the table. |
| e/ | In October 1995, ODE moved its offices from Piccard Drive to Corporate Boulevard in Rockville, Maryland. ODE accepted premarket submissions during the 14-day moving period but added 2 weeks to the due dates of IDEs. This 2-week delay is reflected in the percent of decisions made within the 30 days for original IDEs and amendments. This policy was announced in two notices in the Federal Register of October 14, 1994 (pg. 52170) and November 29, 1994 (pg. 60092). |
FY 96 |
FY 97 |
FY 98 |
FY 99 |
FY 00 |
|
| Number Received | 3,189 | 3,776 | 4,277 | 4,127 | 4,388 |
Number of Decisions |
3,121 |
3,777 |
4,209 |
4,224 |
4,335 |
| Average FDA Review Time (days) | 21 |
21 |
21 |
20 |
20 |
Percent (%) of Decisions Made within 30 Days |
99 |
100 |
100 |
100 |
100 |
Number under Review at End of Perioda |
148 |
216 |
284 |
187 |
239 |
Number Overdue at End of Period |
0 |
0 |
0 |
0 |
0 |
| ______________________ | |
| a/ | The number under review at the end of a period may not reconcile with the number under review at the end of the previous period (plus receipts less approvals) because of deletions and conversions which are not reflected in the table. |
FY 96 |
FY 97 |
FY 98 |
FY 99 |
FY 00 |
|
| Number Originals Received | 5,297 | 5,049 | 4,623 | 4,458 | 4,202 |
| Number of Decisions | |||||
| Substantially equivalent | 4,501 | 4,405 | 3,824 | 3,652 | 3,567 |
| Not substantially equivalent | 64 | 57 | 65 | 66 | 52 |
| Othera | 998 | 693 | 1,340 | 875 | 778 |
| Total | 5,563 | 5,155 | 5,229 | 4,593 | 4,397 |
| Percent(%) not substantially | |||||
| Equivalentb | 1.4 | 1.3 | 1.7 | 1.8 | 1.4 |
| Average Review Time (days) | |||||
| FDA timec | 110 | 97 | 89 | 80 | 77 |
| Total timed | 145 | 130 | 114 | 102 | 102 |
| Median Review Time (days) | |||||
| FDA timec | 85 | 81 | 81 | 71 | 68 |
| Total timed | 88 | 85 | 83 | 76 | 72 |
| Percent (%) of Decisions made within 90 Days, based on |
|||||
| FDA timee | 80 | 95 | 97 | 99 | 100 |
| Total timed | 50 | 58 | 59 | 66 | 66 |
| Number under Review at End of Periodf | |||||
| Activeg | 1,408 | 1,287 | 1,057 | 943 | 850 |
| (Active and overdue) | 0 | 0 | 0 | 0 | 0 |
| On holdh | 821 | 865 | 487 | 461 | 370 |
| Total | 2,229 | 2,152 | 1,544 | 1,404 | 1,220 |
| _______________________ | |
| a/ | Includes final administrative actions that did not result in a substantially equivalent/not substantially equivalent decision because the 510(k) or device/product was: withdrawn by the applicant, deleted due to lack of response, a duplicate, not a device, a transitional device, regulated by CBER, a general purpose article, exempted by regulation, and other miscellaneous actions. |
| b/ | Based on "substantially equivalent" and "not substantially equivalent" decisions only. |
| c/ | FDA time includes all increments of time FDA reviewed a 510(k), so long as the 510(k) document number did not change; changes in 510(k) document numbers occur rarely. |
| d/ | Includes all time from receipt to final decision, i.e., does not exclude time a submission is on hold pending receipt of additional information. |
| e/ | Considers whether FDA review time remained within 90 days, with FDA’s review clock being reset to zero whenever additional information was received (in accordance with 21 CFR 807.87(l)). |
| f/ | The number under review at the end of a period may not reconcile with the number under review at the end of the previous period (plus receipts less decisions) because of deletions and conversions which are not reflected in the table. |
| g/ | FDA responsible for processing notification. |
| h/ | FDA’s processing of notification officially suspended pending receipt of additional information from the submitter. |
Table
15. 510(k) Receipt Cohort Performance*
FY 96 - FY 00
FY 96 |
FY 97 |
FY 98 |
FY 99 |
FY 00 |
||||||
Number of 510(k)s Receiveda |
||||||||||
| Traditional | 5,318 | 5,059 | 4,528 | 3,985 | 2,598 | |||||
| Special | 0 |
0 |
80 | 396 | 456 |
|||||
| Abbreviated | 0 |
0 |
21 | 85 | 104 |
|||||
| Total Receipts | 5,318 |
5,059 |
4,629 | 4,466 | 3,158 |
|||||
| Actions on 510(k)s | ||||||||||
| Substantially Equivalent | 4302 | 4150 | 3569 | 3579 | 2,333 | |||||
| Not Substantially Equivalent(%)b |
57(1.3) |
53(1.3) |
68(1.9) |
60(1.7) |
24(1.0) |
|||||
| Otherc | 959 | 856 | 984 | 777 | 399 | |||||
| Total Actions | 5,318 | 5,059 | 4,621 | 4,416 | 2,756 | |||||
| Average Cumulative Days for 510(k) Decisions Excludes Withdrawals and Deletes |
||||||||||
| FDA Time from Receipt to Final Decisiond |
93 |
91 |
82 |
79 |
61 |
|||||
| Total Time from Receipt to Final Decisione |
120 |
116 |
103 |
100 |
73 |
|||||
| All Decisions Including Withdrawals and Deletes |
||||||||||
| FDA Time from Receipt to Final Decisiond |
91 |
89 |
81 |
78 |
60 |
|||||
| Total Time from Receipt to Final Decisione |
150 |
134 |
116 |
109 |
75 |
|||||
| Number of Decisions (%) within 90 Days, Based on: |
||||||||||
| FDA Days from Receipt to First Action |
4,998 |
(94) |
4,968 |
(98) |
4,612 |
(100) |
4,454 |
(100) |
3,150 |
(100) |
| FDA Cumulative Days from Receipt to |
||||||||||
| Final Decision | 3,472 | (65) | 3,558 | (70) | 3,530 | (76) | 3,367 | (75) | 2,452 | (78) |
| Total Cumulative Days from Receipt to |
||||||||||
| Final Decisione | 2,901 | (55) | 3,025 | (60) | 3,025 | (65) | 2,938 | (66) | 2,214 | (70) |
| Average Number of FDA Cycles from Receipt to Final Action |
1.5 |
1.5 |
1.4 |
1.4 |
1.3 |
|||||
| Percentile FDA (Total) Days from Receipt to Final Action |
||||||||||
| 25th | 51 | (59) | 51 | (57) | 47 | (51) | 41 | (45) | 34 | (41) |
| 50th (Median) | 80 | (88) | 80 | (86) | 75 | (83) | 71 | (78) | 64 | (71) |
| 75th | 115 | (188) | 106 | (175) | 90 | (149) | 90 | (147) | 90 | (123) |
| 90th | 173 | (332) | 172 | (312) | 160 | (256) | 162 | (265) | N/A | (N/A) |
| Number under review as of 9/30/00 |
||||||||||
| Active | 0 | 0 | 0 | 16 | 168 | |||||
| Active and Overdue | 0 | 0 | 0 | 0 | 0 | |||||
| On hold | 0 | 0 | 8 | 34 | 234 | |||||
| Total | 0 | 0 | 8 | 50 | 402 | |||||
| Summary of 510(k) Receipt Cohort |
||||||||||
| Substantially Equivalent | 4,302 | 4,150 | 3,569 | 3,579 | 2,333 | |||||
| Not Substantially Equivalent | 57 | 53 | 68 | 60 | 24 | |||||
| Other | 959 | 836 | 984 | 777 | 399 | |||||
| Under Review | 0 | 0 | 0 | 16 | 168 | |||||
| On Hold | 0 | 0 | 8 | 34 | 234 | |||||
| Total | 5,318 | 5,059 | 4,629 | 4,466 | 3,158 | |||||
| _________________________ | |
| */ | For each fiscal year, September 30, 2000 was used as the cutoff date. The FY00 cohort represents only receipts through June 30, 2000 (first nine months of the fiscal year). |
| a/ | IncludesThird Party 510(k)s: FY97 = 14; FY98 = 18; FY99 = 32; FY00 = 30. |
| b/ | Based on "substantially equivalent" and "not substantially equivalent" decisions only. |
| c/ | Includes final administrative actions that did not result in a substantially equivalent/not substantially equivalent decision because the 510(k) or device/product was: withdrawn by the applicant, deleted due to lack of response, a duplicate, not a device, a transitional device, regulated by CBER, a general purpose article, exempted by regulation, and other miscellaneous actions. |
| d/ | FDA time includes all increments of time FDA reviewed a 510(k), so long as the 510(k) document number did not change; changes in 510(k) document numbers occur rarely. |
| e/ | Includes all time from receipt to final decision, i.e., does not exclude time a submission is on hold pending receipt of additional information. |
ODE is responsible for the program areas through which medical devices are evaluated and cleared for clinical trials and marketing. This Appendix provides summary information about the major programs administered by ODE and includes a brief description of the premarket approval, product development protocol, humanitarian device exemption, investigational device exemption, and premarket notification programs.
Premarket Approval Applications (PMAs)
Under the Federal Food, Drug, and Cosmetic Act (the Act) and the FDA regulations, Code of Federal Regulations, Title 21 (the Regulations), a manufacturer or others must submit a PMA for FDA review and approval before marketing certain new Class III devices. The PMA submitter must provide reasonable assurance that the device is safe and effective for its intended use and that it will be manufactured in accordance with current good manufacturing practices. As part of the review process, FDA may present the PMA to an expert advisory panel for its recommendations. After obtaining the panel recommendations, the agency makes a determination to approve the PMA, deny it, or request additional information. When the FDA either approves or denies the PMA, it must publish a notice in the Federal Register to inform the public of the decision and make available a summary of the safety and effectiveness data upon which the decision is based. This publicly available summary does not include proprietary data or confidential information submitted by the applicant.
Product Development Protocols (PDPs)
The 1976 Medical Device Amendments to the Food, Drug, and Cosmetic Act allowed for two product pathways for a class III device: the PMA or, with prior FDA permission, the notice of completion of a PDP. The PDP process is based upon early consultation between the sponsor and the FDA leading to a device development and testing plan acceptable to both parties. It minimizes the risk that the sponsor will unknowingly pursue — with the associated waste of capital and other resources — the development of a device that FDA will not approve. The PDP plan incorporates four discrete stages of FDA review during the device design process: a PDP Summary Outline; FDA/Advisory Panel review of the full PDP; consideration and, where appropriate, pre-approval of design modifications and protocol revisions made during execution of the PDP; and action on the sponsors Notice of Completion. FDA review of the PDP summary may take up to 30 days; the review of the full PDP may take up to 120 days; and FDA must declare the PDP "completed" or "not completed" within ninety days of receiving the Notice. If the FDA finds that the Notice — together with other information previously submitted — shows that the requirements of the PDP, including Quality
System Regulation Inspection (or GMP inspection in the case of sponsors without an established satisfactory inspection history), have been met, the Agency will declare the PDP complete.
Humanitarian Device Exemptions (HDEs)
An HDE application is essentially the same as a PMA in both form and content but is exempt from the effectiveness requirement of a PMA. Even though the HDE is not required to contain the results of scientifically valid clinical investigations demonstrating that the device is effective for its intended purpose, the application must contain sufficient information for FDA to determine, as required by statute, that the device does not pose an unreasonable or significant risk of illness or injury to patients and that the probable benefit to health outweighs the risk of injury or illness from its use. An HDE application must also contain information that will allow FDA to make the other determinations required by the act. An approved HDE authorizes marketing of the humanitarian use device (HUD).
After a PMA is approved, the PMA holder may request FDA approval of changes to be made. For example, it may request changes to the device, its labeling or packaging, or the manufacturing processes used in its production. Unless prior approval is expressly not required by the PMA regulation, changes that affect the safety or effectiveness of the device require FDA premarket approval. FDA’s review of a PMA supplement may be easy or difficult depending on the type of device, the significance of the change, and the complexity of the technology. Some PMA supplements can be as complex is the original application. Although the statutory timeframe is 180 days for PMA Supplements, FDA is committed to reviewing these in shorter timeframes and has reduced review timeframes through the use of real-time supplement process, 30-day notices, and expedited reviews.
Investigational Device Exemptions (IDEs)
Under the Act and Regulations, an individual, institution or company may sponsor the clinical investigation of a medical device to establish its safety and effectiveness. Before conducting a clinical trial, however, the sponsor must obtain the approval of an institutional review board (IRB) as well as informed consent from the study subjects at the time of their enrollment in the study. If the investigational device study presents a significant risk to the subjects, the sponsor must obtain FDA’s approval of an "investigational device exemption" application (IDE) under 21 CFR 812. The IDE must contain information concerning the study’s investigational plan, report of prior investigations, device manufacture, IRB actions, investigator agreements, subject informed consent form, device labeling, cost of the device, and other matters related to the study. FDA has 30 calendar days from the date of receipt of the application to approve or disapprove an IDE submission.
Although not provided for in the IDE regulations, all submissions related to an original IDE that has been submitted, but not approved, are referred to as "IDE amendments". After an IDE is approved, related submissions are called "supplemental applications" under the regulations. Identification of IDE amendments enables FDA to track each IDE from the time it is originally submitted until the time it is approved.
The IDE regulation requires the sponsor of an investigation of a significant risk device to submit a supplemental application for a number of reasons. For example, a sponsor must submit a supplement if there is a change in the investigational plan when such a change may affect the scientific soundness of the study or the rights, safety, or welfare of the subjects. Supplemental applications also are required for the addition of investigational sites. This regulation also requires the submission of various reports, which are logged in as supplements to IDE applications. These include reports on unanticipated adverse effects of the device; recall and device disposition; failure to obtain informed consent; and annual progress reports, final reports, investigator lists, and other reports requested by FDA.
Premarket Notifications (510(k))
At least 90 days before placing a medical device into commercial distribution, a person required to register must submit to FDA a premarket notification, commonly known as a "510(k)." The exception to this is if the device is exempt from the 510(k) requirements of the Act by statue or regulation. In addition to other information concerning the device, e.g., a description of the device, a 510(k) summary or a 510(k) statement, the 510(k) submitter must include information to substantiate that the device is "substantially equivalent" to a legally marketed device that is not subject to premarket approval. A substantially equivalent device is marketed subject to the same regulatory controls as the device to which it is found to be substantially equivalent. A device may not be marketed pursuant to a 510(k) until the submitter receives written clearance from FDA.
The following is a bibliography of articles and abstracts prepared by the ODE staff and published or presented during FY 2000.
Journals, Newsletter Articles and Book Chapters
Atwood CS, Hovey RC, Glover JP, Chepko G, Ginsburg E, Robison WG Jr. and Vonderhaar BK. Progesterone Induces Side Branching of the Ductal Epithelium in the Mammary Glands of Peripubertal Mice. J. Endocrinol. 167(1): 39-52, 2000.
Banu N, Mozes MM, Kopp JB, Ziyadeh FN, and Meyers CM. Regulation of Inducible Class II
MHC, Costimulatory Molecules, and Cytokine Expression in TGF-![]() 1 Knock-Out Renal Epithelial Cells.
Effects of Exogenous TGF-beta1. Journal of the American Society of Nephrology
10:507a, 1999.
1 Knock-Out Renal Epithelial Cells.
Effects of Exogenous TGF-beta1. Journal of the American Society of Nephrology
10:507a, 1999.
Carey CC and Callahan TJ. Automated External Defibrillators in Children: Food and Drug Administration Issues. In Ventricular Fibrillation: A Pediatric Problem, edited by Linda Quan and Wayne H. Franklin, Armonk, NY: Futura Publishing Company, Inc., pp 195-208, 2000.
Carpenter CF and Ticehurst JR. Non-A, Non-B, or Non-C Hepatitis. Current Treatment Options in Infectious Diseases 2(5):423-429, 2000.
Chenault VM and Benson CC. Regulatory Aspects of Lipid and Lipoprotein Measurements. In Handbook of Lipoprotein Testing, 2nd Edition, edited by Nader Rafai, G. Russell Warnick and Marek H. Dominiczak, American Association for Clinical Chemistry Press, pp 767-794, 2000.
Cornelius MJ. FDA Guidelines for Endoscope Reprocessing. Gastrointestinal Endoscopy Clinics of North America 10(2):259-264, 2000.
Glover JP, Jacot JL, Basso MD, Hohman TC, and Robison WG Jr. Retinal Capillary Dilation: Early Diabetic-Like Retinopathy in the Galactose-Fed Rat Model. Journal of Ocular Pharmacology and Therapeutics 16(2):167-172, 2000.
Gutman S and Richter K. New Directions in the FDA Regulation of In Vitro Diagnostic Devices. Laboratory Medicine 30(12):782-785, 1999.
Hackett JL. FDA Takes Over CLIA Complexity Determinations. IVD Technology 6(3):26-28, 2000.
Hellman KJB, Solomon RR, Gaffey C, Durfor CN, and Bishop JG. Regulatory Considerations. In Principles of Tissue Engineering, 2nd edition, edited by R.P. Lanza, R. Langer and J. Vacanti, San Diego, Academic Press, pp 915-927, 2000.
Hirschl R and O’Neill C. Regulatory Issues Related to Extracorporeal Life Support. In ECMO: Extracorporeal Cardiopulmonary Support in Critical Care, 2nd edition, edited by J.B. Zwischenberger, R.H. Steinhorn, and R.H. Bartlett, Ann Arbor, Extracorporeal Life Support Organization, pp 701-706, 2000.
Houn F, Bright RA, Bushar HF, Croft BY, Finder CA, Gohagan JK, Jennings RJ, Keegan P, Kessler LG, Kramer BS, Martynec LO, Robinowitz M, Sacks WM, Schultz, DG, and Wagner RF. Study Design in the Evaluation of Breast Cancer Imaging Technologies. Acad. Radiol. 7:684-92, 2000.
McCullagh L and Baker KH. Endoscope Reprocessing: Taking the Mystery out of HIgh-Level Disinfection. ORL - Head and Neck Nurses 18:1:6-10, Winter 2000.
Moxey-Mims MM, Young G, Silverman A, Selby D, White JG, and Kher KK. End-Stage Renal Disease in Two Pediatric Patients with Fechtner Syndrome. Pediatr. Nephrol. 13(9):782-786, 1999.
Ng CS and Rosenthal AR. Classification & Epidemiology of Uveitis. In Oxford Textbook of Ophthalmology, edited by D.L. Easty and J.M. Sparrow, Oxford University Press, Vol. 1, pp 509-515, 1999.
Ng CS and Rosenthal AR. Principles of Clinical Management of Uveitis. In Oxford Textbook of Ophthalmology, edited by D.L. Easty and J.M. Sparrow, Oxford University Press, Vol. 1, pp 553-559, 1999.
Robison WG Jr., Cook-Ashby JC, Soto I, Kelley MA, Glover JP, and Jacot JL. Identification of Vasculogenic-Like Precursor Cells in the Galactose-Fed Rat Model of Diabetic Retinal Microangiopathies. Investigative Ophthalmology and Visual Science 41(4):S405, 2000.
Robison WG Jr., Jacot JL, Katz ML, and Glover JP. Retinal Vascular Changes Induced by the Oxidative Stress of a -Tocopherol Deficiency Contrasted with Diabetic Microangiopathy. Journal of Ocular Pharmacology and Therapeutics 16(2):109-120, 2000.
Sauberman HR. Food and Drug Administration Approval Process for Cochlear Implants. In Cochlear Implants, Principles and Practices, Appendix 6B; edited by John K. Niparko, M.D., Johns Hopkins University, Lippincott Williams & Wilkins, Philadelphia, pp 122-128, 2000.
Spyker DA, Harvey ED, Harvey BE, Harvey AM, Rumack BH, Peck CC, Atkinson AJ Jr., Woosley RL, Abernethy DR, and Cantilena LR. Assessment and Reporting of Clinical Pharmacology Information in Drug Labeling. Clin. Pharmacol. Ther. 67(3):196-200, 2000.
Abstracts and Presentations
Balow J and Meyers CM. Iatrogenic Nephrotic Syndrome (Graft-vs-Host Disease and Renal Involvement), Federal Medical Monthly Nephrology Seminars, Uniformed Services University of the Health Sciences, Bethesda, MD, March 2000.
Banu N, Mozes MM, Kopp JB, Ziyadeh FN, and Meyers CM. Regulation of Inducible Class II
MHC, Costimulatory Molecules, and Cytokine Expression in TGF-![]() 1 Knock-Out Renal Epithelial Cells.
Effects of exogenous TGF-
1 Knock-Out Renal Epithelial Cells.
Effects of exogenous TGF-![]() 1. American Society of Nephrology Meeting, Miami, FL,
November 1999.
1. American Society of Nephrology Meeting, Miami, FL,
November 1999.
Baker KH and Cygnarowicz T. A Multidisciplinary Approach to Development of a Guidance Document for Implantable Middle Ear Hearing Devices. FDA Science Forum in Washington D.C., February 2000.
Baker KH and McCullagh L. Glove Selection: Choosing the Right Glove for the Job. Society of Otolaryngology/Head and Neck Nurses Annual Congress and Symposium, Washington, D.C., September 2000.
Boulware A, Eydelman MB, Lum F, Silverman P, and Lochner D. Retrospective Evaluation of IOLs in Adults Under 60 Years. Symposium on Cataract, IOL and Refractive Surgery, American Society of Cataract and Refractive Surgery, Boston, MA, May 2000.
Calogero D, Eydelman MB, Arshinoff S, Senft S, Bilotta M, and Hadi H. Endothelial Cell Loss with Viscoelastic Use. Symposium on Cataract, IOL and Refractive Surgery, American Society of Cataract and Refractive Surgery, Boston, MA, May 2000.
Carey CC and Saperstein W. Testing Requirements for Automated External Defibrillators (AEDs). FDA Science Forum, Washington, D.C., February 2000.
Demian, H. A Regulatory Perspective for Orthopedic Bone Cements. 6th World Biomaterials Congress, Kamuela, Hawaii, May 2000.
Eydelman MB, Calogero D, Arsinoff S, Senft S, Bilotta R, and Hadi H. Development of an International Standard for the Clinical Evaluation of New Viscoelastics. American Society of Cataract and Refractive Surgery Annual Meeting, Boston, MA, May 2000.
Forman M, Oberste MS, Ray SC, Pallansch M, and Ticehurst J. Parechovirus Detection by PCR: Interstrain Heterogeneity and Optimization. 16th Annual Clinical Virology Symposium and Annual Meeting, Pan American Society for Clinical Virology, Clearwater Beach, FL, April - May 2000.
Fugate KJ, Vadlamudi S, Turkeltaub P, and Noah C. Development of an In-Vitro IgE Test for Ethylene Oxide Hypersensitivity: A CRADA Study and Technology Transfer Agreement with Diagnostic Product Corporation, Inc., National Institutes of Health and Centers for Disease Control and Prevention. FDA Science Forum, Washington, D.C., February 2000.
Gatling, RR. Review of Various PMA Approaches. AdvaMed's Tenth Annual Device Submissions Workshop, Washington, D.C., July 2000.
Gatling, RR. Modifications to Devices Subject to Premarket Approval - The PMA Supplement Decision Making Process. AdvaMed's Tenth Annual Device Submissions Workshop, Washington, D.C., July 2000.
Gatling, RR. 30-Day Notices and 135-Day PMA Supplements for Manufacturing Changes. AdvaMed's Tenth Annual Device Submissions Workshop, Washington, D.C., July 2000.
Gatling, RR. Least Burdensome Provisions of FDAMA. 11th Annual Medical Device Technology European Conference, Paris, France, September 2000.
Harvey B. The Place of Virtual Bronchoscopy in Clinical Practice: Barriers and Solutions. International Society for Optical Engineering (SPIE), Medical Imaging 2000, San Diego, CA, February 2000.
Harvey B. FDA Premarket Regulatory Requirements for In Vitro Diagnostic Devices. Health Care Financing Administration (HCFA) Laboratory & Diagnostic Services Panel, Public Advisory Panel Meeting, Baltimore, MD, November 1999.
Harvey B. How the FDA, Industry and Clinicians Deal with New Medical Information. American College of Gastroenterology Ad Hoc Committee on FDA Related Matters, 64th Annual Scientific Meeting, American College of Gastroenterology, Phoenix, AZ, October 1999.
Harvey B. The Regulation of Medical Devices by the U.S. Food and Drug Administration: the Rest of the Story. Gastroenterology Grand Rounds, University of Virginia School of Medicine, Charlottesville, VA, October 1999.
Harvey E, Mitchell D, and Schultz D. Preclinical Animal and Clinical Testing Guidelines for New Adhesion Barriers, 4th International Conference on Postoperative Healing, Ft. Lauderdale, FL, October 1999.
Ho C. Assessment of the Shelf Lives of Cardiac Devices. FDA Science Forum, Washington, D.C., February 2000.
Horbowyj R. Publishing in International Medical Literature. World Federation of Ukranian Medical Associations, VIII Congress, Lviv, Ukraine, August 2000.
Jevtich MJ. FDA Experience with Modern BPH Devices and Regulatory Process. 5th International Consultatio on BPH (WHO), Paris, France, June 2000.
Kammula R. Use of FDA Recognized Standards and Master Files to Address Biocompatibility. Medical Design and Manufacturing (MD&M) East Conference, New York City, NY, June 2000.
Maxim PE. Pharmacogenomics: A Regulatory Perspective. The Second Annual Pharmacogenomics Event, London, England, January 2000.
Maxim PE. FDA Review of In Vitro Medical Devices (informal presentation to staff). Medical Devices Agency, London, England, January 2000.
Maxim PE. Performance Studies for Allergen Reagents. Technical Consultant Meeting on IgE Allergy Testing, CDC, Atlanta, GA, April 2000.
Maxim PE. FDA and Genetic Testing. HCFA-RO Laboratory Conference, Baltimore, MD, May 2000.
Maxim PE. FDA Regulation of In Vitro Medical Devices. Tri-Service Clinical Investigation Postgraduate Short Course, San Antonio, TX, May 2000.
Maxim PE. Clinical Trials: What the FDA’s New IVD Policy Means to You, AACC Teleconference, Rockville, MD, June 2000.
Maxim PE. FDA and Genetic Testing: An Update on SACGT Proceedings. Professional Roundtable, Rockville, MD, June 2000.
Maxim PE. Role of International Standards and Reference Materials in FDA Regulation of IVDs. WHO Consultation on International Biological Standards, Geneva, Switzerland, September 2000.
Maxim PE. Data Requirements for 510(k) Submissions to DCLD. AMDM In Vitro Diagnostics 510(k) Workshop, Rockville, MD, September 2000.
Melkerson M. To Evaluate Why, What and Wherein Orthopedic Device Standards: Planning for 2000 and Beyond. American Society for Testing and Materials, Toronto, Canada, May 2000.
Meyers CM and Jevnikar A. Breakthroughs and Hot Topics in Cellular Immunity. Scientific Session Moderators. American Society of Nephrology Meeting, Miami, FL, November 1999.
Michaud G. Quality Control of Point of Care Coagulation Devices. The 46th SSC Meeting of the ISTH, Maastricht, The Netherlands, June 2000.
Michaud G. Novel Designs with Traditional Names. When Should an Assay be Given a New Name? The 46th SSC Meeting of the ISTH, Maastricht, The Netherlands, June 2000.
Morris JM. Regulatory Considerations for Emerging Surface Technology, Surfaces in Biomaterials 2000, Scottsdale, AZ, September 2000.
Neuland CY. Requesting Evaluation of an Automatic Class III Designation. HIMA Workshop: Getting to Market Sooner, Washington, D.C., December 1999.
Pollard CM. In Vivo Diagnostic Spectroscopy: Regulatory Considerations for Clinical Trials and Premarket Clearance, Optical Society of America, Miami Beach, FL, April 2000.
Poole FM. FDA Requirements for Microbiological Specimen Transport System. NCCLS Subcommittee for Quality Control of Microbiological Transport Systems, February 2000.
Rechen, EJ. Third-Party Review of 510(k)s. HIMA Workshop: Getting to Market Sooner, Washington, D.C., December 1999.
Rechen, EJ. Third Party Review. AdvaMed's Tenth Annual Device Submissions Workshop, Washington, D.C., July 2000.
Rechen, EJ. FDA's 510(k) Third-Party Review Program, CBER Blood Products Advisory Committee Meeting, Gaithersburg, MD, September 2000.
Rechen, EJ. FDA's 510(k) Third-Party Review Program, FDA Medical Device Workshop, Depew, NY, September 2000.
Robison WG Jr., Glover JP, Jacot JL, Basso MD, Hohman TC. ARI Prevention of Retinal Capillary Dilation, an Early Diabetic-Like Microangiopathy in the Galactose-Fed Rat Model. US-Japan Aldose Reductase Workshop, Kona, Hawaii, January 2000.
Robison WG Jr., Jacot JL, Katz ML, Glover JP. Relative Roles of Oxidative Stress and Elevated Aldose Reductase Activity in the Microangiopathies of Diabetic Retinopathy. US-Japan Aldose Reductase Workshop, Kona, Hawaii, January 2000.
Rosenthal AR and Eydelman MB. A Guide to Ophthalmic Device Evaluation. American Academy of Ophthalmology, Orlando, FL, October 1999.
Schultz D. Technology Assessment, An FDA Perspective. American College of Surgeons Annual Clinical Conference, San Francisco, CA, October 1999.
Shively RG. Design Issues for Nucleic Acid Amplification Testing. TB Task Force Meeting, NIH, Bethesda, MD, December 2000.
Shulman, M. Premarket Notification (510(k)s). Dallas District FDA/Industry Medical Device Coalition, Dallas, TX, October 1999.
Shulman, M. Implementation of the 510(k) Paradigm and Premarket Notification. Medical Design and Manufacturing (MD&M) Minneapolis, MN, October 1999.
Shulman, M. Premarket Notification and FDA CDRH Hot Topics for 2000. Medical Design and Manufacturing (MD&M) East Conference, New York City, NY, June 2000.
Shulman, M. Premarket Notification Regulatory Review. AMDM In Vitro Diagnostics 510(k) Workshop, Rockville, MD, September 2000.
Ticehurst J. Evidence Based Diagnostic Virology. 16th Annual Clinical Virology Symposium and Annual Meeting, Pan American Society for Clinical Virology, Clearwater Beach, FL, April - May 2000.
Ticehurst J. FDA Approaches to Assays for Diagnosis or Monitoring of HCV Infections. Association of Public Health Laboratories Conference on Laboratory Aspects of Human Retrovirus & Hepatitis C Testing, Charlotte, NC, March 2000.
Whitaker KB, Shively RG, and Dubois W. CDRH Perspective on Nucleic Acid Amplification Testing. FDA/Industry Training Co-sponsored by FDA and Gen-Probe, Inc., Roche Molecular Systems, Chiron/Bayer Diagnostics, and Organon Teknika, May 2000.
Zhou S and Hoang Q. Test for Comparability of Excimer Lasers. FDA Science Forum, Washington, D.C., February 2000.
Staff College Presenters and Faculty
| Aziz, Kaiser | Less, Joanne | Rechen, Eric |
| Beers, Everette | Melvin, Marsha | Romanell, Lawrence |
| Brown, Daniel W.C. | Mishra, Nirmal | Rosecrans, Heather |
| Durfor, Charles | Morris, Janine | Shulman, Marjorie |
| Gantt, A. Doyle | Neuland, Carolyn | Sliva, Clara |
| Goode, Jennifer | Nutter, Cathy | Tillman, Donna-Bea |
| Gutman, Steve | Nguyen, Trinh | Turtil, Steven |
| Horbowyj, Roxolana | Phillips, Robert | Ulatowski, Tim |
| Kammula, Raja | Phillips, Philip | Weitershausen, Joanna |
| Kennell, Lisa | Poneleit, Kathy | Witten, Celia |
| Lacy, Frank | Portnoy, Stuart | Zuckerman, Bra |

Office of the Director |
Division of Dental, Infection Control, and General Hospital Devices (Continued) |
Acker, Rita |
Foster, Sarah |
| Cooper, Brooksie | Fox, Pat |
| DeMarco, Carl | Fuller, Janie |
| Gibbs, Danielle | Hibbard, Viola |
| Gornick, MaryAnn | Hoard, Renita |
| Hobbs, Cathy | Levchuck, John |
| Phillips, Philip | Lin, Chiu |
| Pluhowski, Nancy | Marshall, Felicidad |
| Poneleit, Kathy | Mayhall, Elaine |
| Richter, Kimber | Naveau, Irene |
| Sauberman, Harry | O’Connell, Linh |
| Schultz, Dan | O’Lone, Martha |
| Statland, Bernard | Robinson, Mary Jo |
Program Management Office |
Runner, Susan Samuels-Reid, Joy |
Appler, Kathryn |
Scott, Pam Shipps, Gerald |
| Broughton, Shirley | Shire, Sandra |
| Cancino, Isella | Smith, Gwen |
| Clingerman, Angie | Soprey, Pandu |
| Dowtin, Lesa | Turtil, Steve |
| Dumas, Evalee | Ulatowski, Tim |
| Howell, Kimberly Jaeger, Jeff Koviack, Bob |
Division of General, Restorative, and Neurological Devices |
| Robins, Lisa Schielke, Mary |
Allen, Peter |
| Wedlock, Chuck | Allen, Samie |
| Wilson, Robin | Anderson, Jodi |
Program Operations Staff |
Arepalli, Sam Basu, Sankar |
Berk, Gene |
Berkowitz, David Bernato, Delores |
| Fisher, Lisa | Berne, Bernard |
| Gatling, Robert | Biddle, Timothy |
| Less, Joanne | Blair, Therian |
| Lyons, Linda | Bourke, Tracey |
| Melvin, Marsha | Bowsher, Kristen |
| Nguyen, Thinh | Costello, Ann |
| Parker, Mervin | Courtney, Mike |
| Perticone, Diane | Dawisha, Sahar |
| Rechen, Eric | DeLuca, Bob |
| Rosecrans, Heather | Demian, Hany |
| Sawyer-Major, Wanda | Durfor, Charles |
| Shulman, Marjorie | Einberg, Elmar |
| Williams, Paul | Eudy, Mike |
| Wolanski, Nicole | Felten, Richard |
Division of Clinical Laboratory Devices |
Fogarty, Pauline Foy, Keith |
Aziz, Kaiserx |
Gadaleta, Sergio Gantt, Gail |
| Bautista, Josephine | Goode, John |
| Benson, Carol | Hammond, Della |
| Bernhardt, Pat | Hinckley, Steve |
| Blagmon, Djuana | Horbowyj, Roxi |
| Brindza, Larry | Hudson, Peter |
| Bucher, Betty | Kaiser, Aric |
| Callaghan, Jim | Keith, Erin |
| Calvin, Veronica | Kim, Sam |
| Chace, Nina | Krause, David |
| Chan, Maria | Lee, Kevin |
| Chenault, Michelle | Mattamal, George |
| Chesler, Ruth | Mattera, Michelle |
| Clark-Stuart, Michelle | Melkerson, Mark |
| Cooper, Jean | Mishra, Nirmal |
| Dada, Valerie | Morris, Janine |
| Danishefsky, Avis | Ogden, Neil |
| Diggs, Denise | Pagano, Russell |
| Dubois, Woody | Pak, Yung |
| Fourcroy, Jean | Phillips, Mary Ellen |
| Fugate, Kearby | Rhodes, Holly |
| Gaffey, Claudia | Rhodes, Stephen |
| Gonzalez, Augustin | Schroeder, Marie |
| Gutierrez, Alberto | Scudiero, Jan |
| Gutman, Steve | Sloan, Nadine |
| Hackett, Joe | Stevens, Ted |
| Hanna, Nancy | Stiegman, Glenn |
| Hawthorne, Ann | Sturniolo, Mike |
| Heyliger, Marian | Sung, Pei |
| Hyde, John | Teresinski, Doris |
| Ingram, Kenneth | Torres-Cabassa, Angel |
| Jones, Doris | Tudor, Natalie |
| King, Lisa | Warfield, Diana |
| Lyle, Dave | Watson, Tony |
| MacArthy, Philip | Weiblinger, Rick |
| Magruder, Louise | Witten, Celia |
| Maxim, Peter | Wolf, Beverly |
| McClain-Bennett, Joan | Yahiro, Martin |
| Michaud, Ginette | Yen, Dwight |
| Moore, Deborah Moxey-Mims, Marva Peacock, Albert |
Division of Ophthalmic and Ear, Nose, and Throat Devices |
| Pinkos, Arleen Poole, Freddie |
Alexander, Kesia |
| Radha, Edappallath | Baker, Karen |
| Rao, Prasad | Beers, Everette |
| Reeves, Pat | Berman, Sheryl |
| Robinowitz, Max | Boulware, Ashley |
| Rogers, Liz | Brogdon, Nancy |
| Selepak, Sally | Brown, Daniel |
| Shively, Roxanne | Burke-Nicholas, Marsha |
| Simms, Tom | Callaway, Jan |
| Sliva, Clara | Calogero, Don |
| St. Pierre, Don | Chen, Tzeng |
| Summers, Peter | Cohen, Linda |
| Ticehurst, John | Cygnarowicz, Teresa |
| Tsai, Miin-Rong | Drum, Bruce |
| Vadlamudi, Kris | Eydelman, Malvina |
| Weeks, Susan | Falls, Deborah |
| Wei, Tena | Felton, Eleanor |
| Whitaker, Kathleen | Glover, Joel |
| Wilbon, Tonya | Gouge, Susan |
| Wood, Geretta | Hilmantel, Gene |
| Wright, Kathy | Hoang, Quynh |
Division of Cardiovascular and Respiratory Devices |
Jaffe, Sidney Jones, Susanna Kane, James |
Abel, Dorothy |
Kaufman, Daryl Krawczyk, Claudine |
| Bazaral, Mike | Lepri, Bernard |
| Berman, Mike | Leslie, Sharmeka |
| Brown, Michele | Lochner, Donna |
| Buckley, Donna | Malshet, Vasant |
| Callahan, Tom | McCarthy, Denis |
| Carey, Carole | Montgomery, Al |
| Chandeysson, Paul | Moore, Shirley |
| Cheng, Jim | Ortega, Maritze |
| Ciarkowski, Art | Romanell, Jake |
| Danielson, Judy | Rorer, Eva |
| Demian, Cindy | Rosenthal, Ralph |
| Dillard, Jim | Saviola, James |
| Donelson, Jan | Selfon, Eric |
| Fleischer, Dina | Sharpe, Skip |
| Foreman, Christy | Shi, Dexiu |
| Foster, Elaine | Shih, Ming-Chuen |
| Foy, Joni | Smith, Myra |
| Gabriel, Lynette | Storer, Patricia |
| Gantt, Doyle | Thornton, Sara |
| Gibbons, Gwen | Toy, Jeffrey |
| Gomez-Novoa, Carmelina | Warburton, Karen |
| Goode, Jennifer | Waxler, Morris |
| Ho, Charles | Whipple, David |
| Hottenstein, Omar Huynh, Ann Hwang, Shang |
Division of Reproductive, Abdominal, and Radiological Devices |
| Jensen, Nick Jones, Edwena |
Allen, Cheryl |
| Kaiser, Suzanne | Arnaudo, Joe |
| Karanian, John | Baxley, John |
| Kennell, Lisa | Byrd, Laura |
| Kroen, Marian | Chen, John |
| Kurtzman, Steve | Cooper, Jeff |
| Lacy, Frank | Cornelius, Mary Jo |
| Lacy, Fred | Corrado, Julia |
| Lee, James | Czerska, Ewa |
| Lemperle, Bette | Dart, Linda |
| Letzing, Bill | Daws-Kopp, Kathryn |
| Lyle, Judy | Doyle, Bob |
| Mazzaferro, Bob | Eba, Felissa |
| Moynahan, Megan | Fredericksen, Jane |
| Nakayama, Von | Gammell, Paul |
| Nell, Diane | Gonzalez, Gema |
| Noe, William | Harvey, Brian |
| Oktay, Semih | Harvey, Elisa |
| O’Neill, Carroll | Herrera, Hector |
| Parkhurst, John | Jevtich, Milorad |
| Peters, Kimberly | Kammula, Raju |
| Portnoy, Stuart | Kang, Andrew |
| Puglisi, Mike | Kuchinski, Mike |
| Roy, Joydeb | Lappalainen, Sharon |
| Ryan, Tara | Lawrence, Lisa |
| Sapirstein, Wolf | Lutwak, Leo |
| Shanker, Rhona | Mackey, Cheryl |
| Shein, Mitch | Mallis, Elias |
| Sloan, Chris | McCool, Barbara |
| Smallwood, Senora | McGee, Leah |
| Stuhlmuller, John | Meyers, Catherine |
| Subramanian, Ramiah | Miller, Linda |
| Terry, Doris | Miller, Pat |
| Tillman, Donna-Bea | Mitchell, Diane |
| Usher, Will | Monahan, Jack |
| Wang, Emil | Neuland, Carolyn |
| Weitershausen, Joanna | Nimmagadda, Rao |
| Wentz, Catherine | Nutter, Cathy |
| Zimmerman, Barbara | O’Brien, Mary Beth |
| Zuckerman, Bram | Olvey, Kathleen |
Division of Dental, Infection Control, and General Hospital Devices |
Perez, Rod Phillips, Bob Pollard, Colin |
Adjodha, Michael |
Price, Veronica Provost, Miriam |
| Barrett, Sue | Rubendall, Rita |
| Betz, Robert | Sacks, William |
| Bexabeh, Shewit | Sauls, Mattie |
| Blackwell, Angela | Segerson, Dave |
| Blount, Sharon | Seiler, Jim |
| Bolden, Brenda | Shuping, Ralph |
| Browne, Myra | Virmani, Mridulika |
| Burdick, William | Williams, Dick |
| Cricenti, Pat | Whang, Joyce |
| Cunningham, Terrell | Zaremba, Loren |
| Dorsey, Regina | Zaudtke, Peter |
| Floyd, Chirelle |
Updated 4/9/2001
![]()
CDRH Home Page | CDRH A-Z Index | Contact CDRH | Accessibility | Disclaimer
FDA Home Page | Search FDA Site | FDA A-Z Index | Contact FDA | HHS Home Page
Center for Devices and Radiological Health / CDRH

