|
La provisión completa de ADN-todos los genes y espacios entre ellos-en todos los cromosomas de una especie se conoce como su genoma. Con excepción de los glóbulos rojos, los cuáles no tienen núcleo, el genoma humano está localizado en el núcleo de cada célula del cuerpo. Ahí se encuentra organizado en 46 moléculas muy grandes conocidas como cromosomas; 44 se conocen como autosomas y 2 se conocen como los cromosomas sexuales.
Una colaboración internacional conocida como el Proyecto del Genoma Humano (Human Genome Project, en inglés) ha identificado cada base química en el genoma humano y ha descubierto que hay alrededor de 25,000 genes presentes.

La genómica del cáncer es el estudio del genoma de cáncer humano. Es una búsqueda dentro de las "familias con cáncer" y los pacientes para la recolección completa de genes y mutaciones-tanto heredadas como esporádicas-que contribuyen al desarrollo de una célula cancerosa y su progresión de un cáncer localizado a uno que crece sin control y se metastatiza (se disemina por todo el cuerpo humano).
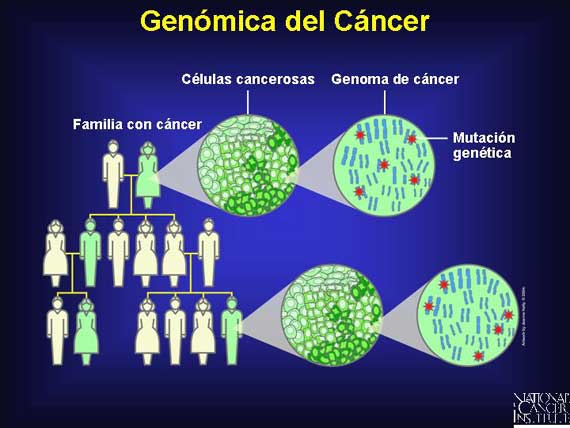
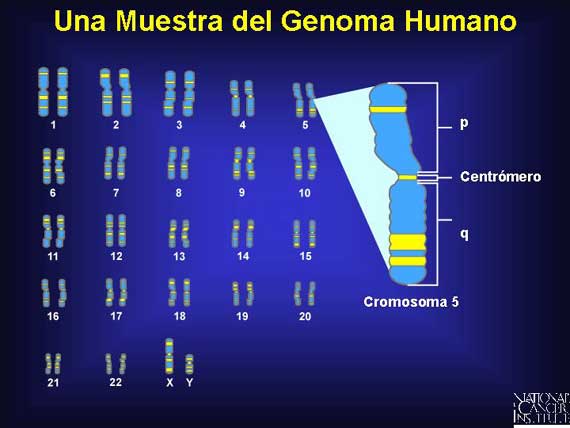
Un cariotipo humano es una exhibición de su genoma. Muestra todos los cromosomas presentes en un individuo después de que se hayan teñido y arreglado en pares conocidos como homólogos. Éste es un cariotipo masculino debido a que hay un cromosoma X y uno Y presentes.
El centrómero de un cromosoma es la región que separa los dos brazos. El brazo que se encuentra arriba del centrómero, el cuál es el más corto, se conoce como el brazo p, mientras que el brazo más largo es el brazo q.
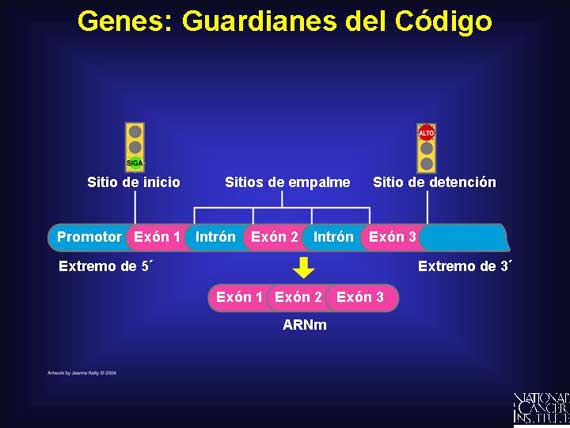
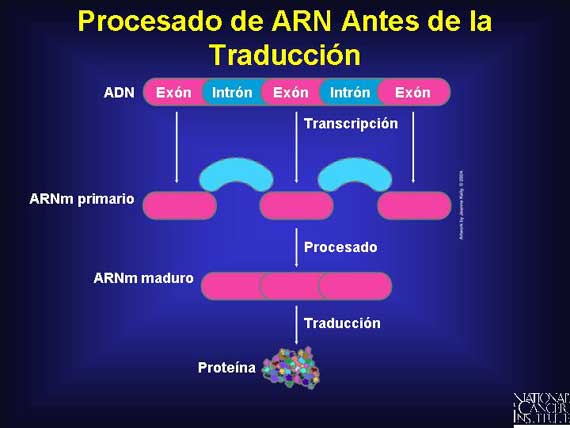
Los 25,000 genes esparcidos por todos los cromosomas humanos incluyen alrededor de sólo un 3 por ciento del genoma total. Estos genes contienen información que es sumamente importante para toda la vida humana. Aunque todas las bases de componente en un gen se copian a medida que la información sale del núcleo, no toda esta información es conservada. Esto se debe a que dentro de un gen hay segmentos de bases tanto codificados como no codificados. Por ejemplo, en los genes divididos, las secciones codificadas conocidas como exones proporcionan las instrucciones genéticas que son copiadas para dirigir la síntesis de proteínas. Estas secciones se preservan pero otras secciones no codificadas dentro del gen, conocidas como intrones, son removidas y degradadas rápidamente.
Cerca de cada gen se encuentra una secuencia "reguladora" de ADN, la cual es capaz de "activar" o "desactivar" al gen. Algo más lejos hay regiones potenciadoras o amplificantes, las cuales pueden acelerar la actividad de un gen.
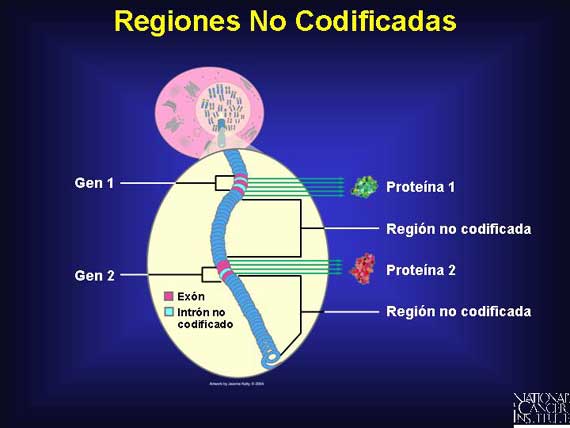
Las moléculas del ADN masivas conocidas como cromosomas también tienen muchas regiones no codificadas localizadas fuera de los genes. Estas regiones contienen segmentos grandes de secuencias repetitivas. Algunas de las secuencias en estas localizaciones están involucradas en la regulación de la expresión genética y otras simplemente actúan como espaciadores. Y todavía hay otras regiones que tienen funciones que aún no se han descubierto.
Cuando un gen "se activa", eventualmente produce una proteína, pero no lo hace así directamente. Primero, el gen codifica a una molécula intermediaria conocida como ARNm. Para transferir la información del gen desde el ADN al ARNm, se utiliza el apareamiento de bases. Sin embargo, hay un cambio: Una base de adenina (A) en el ADN se aparea con una nueva base conocida como uracilo (U) en el ARNm. Esta diferencia ayuda a distinguir al ARNm del ADN.
El ARNm viaja desde el núcleo hacia el citoplasma a organelos celulares conocidos como ribosomas. Ahí, dirige el ensamblado de aminoácidos que se pliegan en una proteína única.
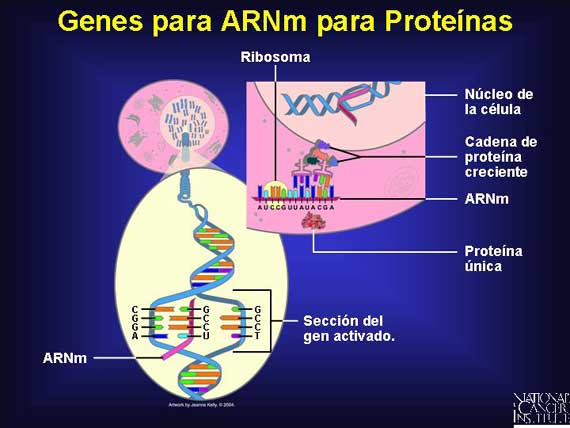
Antes de que el ARNm salga del núcleo, experimenta un procesado adicional. Las regiones no involucradas en la síntesis de proteínas, conocidas como intrones, son suprimidas del mensaje. El ARN maduro que llega a un ribosoma contiene sólo exones que se utilizarán para sintetizar una proteína en un proceso conocido como traducción.
 |
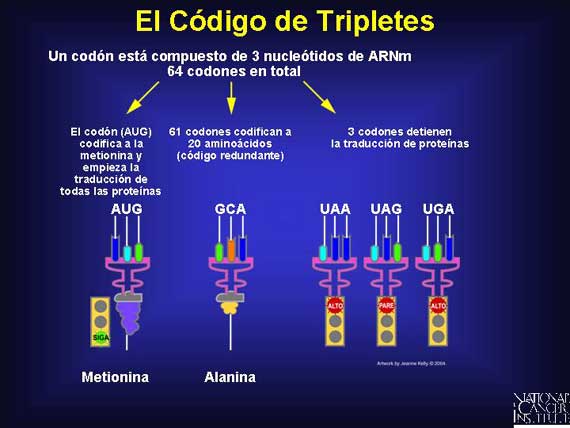
La traducción de las secuencias de bases del ADN a proteína es dependiente del triplete de nucleótidos en el ARNm. Cada triplete de nucleótidos en el ARNm, conocido como un codón, codifica un aminoácido individual y, finalmente, una cadena de aminoácidos forma una proteína. Debido a que el ADN complementario que especifica a un ARNm en particular tiene sólo cuatro bases de nucleótido en un gen, 64 (4X4X4) combinaciones posibles de codones están disponibles para codificar 20 aminoácidos. Así que hay mucha redundancia. Hay 60 tripletes de ARNm para 19 aminoácidos, 3 tripletes para "parar' o "detener" y 1 triplete para expresar a la metionina, el 20avo. aminoácido, que señala "iniciar". La mayoría de los aminoácidos son codificados por más de un codón triplete. Sin embargo, cada triplete está ligado a sólo un aminoácido. (Para obtener información adicional acerca de cómo los genes sintetizan proteínas, por favor, consulte la sección "Genetic Variation" lo que significa en español "Variación Genética").
|
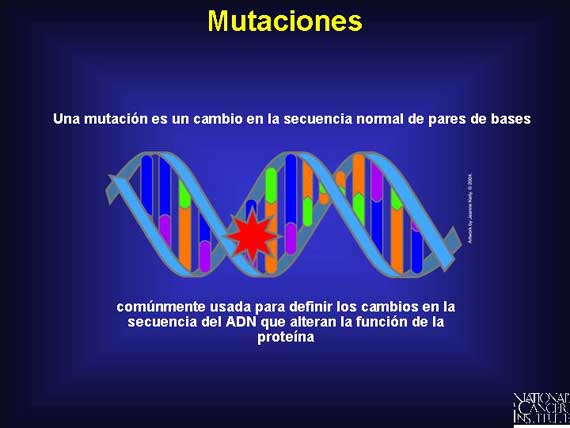
Todas las mutaciones son cambios en la secuencia normal de bases del ADN. Estos cambios pueden ocurrir ya sea en las regiones codificadas o no codificadas. Las mutaciones pueden ser silenciosas y no tener efecto alguno en la proteína resultante. Esto es especialmente cierto si ellas ocurren en regiones no codificadas del ADN. Sin embargo, aún los cambios en pares de bases en la región codificada pueden ser silenciosos debido a la redundancia del código. Por ejemplo, puede ocurrir una mutación dentro de un codón, y sin embargo aún expresar el mismo aminoácido que se había expresado anteriormente.
Las mutaciones pueden involucrar un cambio de base individual-conocido como una mutación de punto-o pueden involucrar a secciones más grandes de ADN a través de supresiones, inserciones o translocaciones.
|
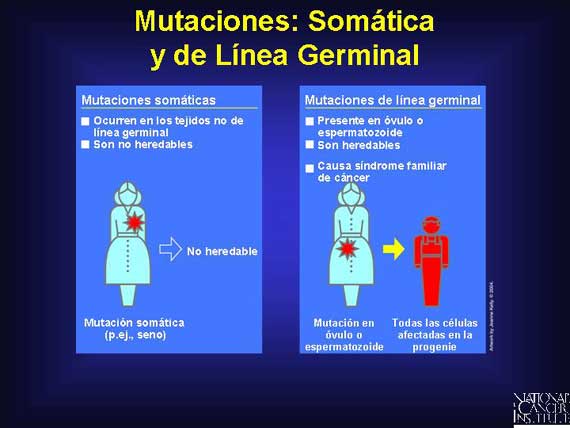
La mayoría de los cánceres se originan en varias mutaciones genéticas que se acumulan en las células del cuerpo durante el periodo de vida
de una persona. Éstas se conocen como mutaciones somáticas y los genes involucrados por lo general están localizados en autosomas
(cromosomas no sexuales). El cáncer también puede tener un componente de mutación de línea germinal, lo
que significa que ellos ocurren en células germinales, mejor conocidas como óvulo o espermatozoide.
Las mutaciones de línea germinal pueden ocurrir de novo (por primera vez) o ser heredadas de las células germinales de los padres. Un ejemplo de mutaciones de línea germinal enlazadas al cáncer son aquéllas que ocurren en los genes de susceptibilidad al cáncer, incrementando el riesgo de una persona para desarrollar la enfermedad.
|
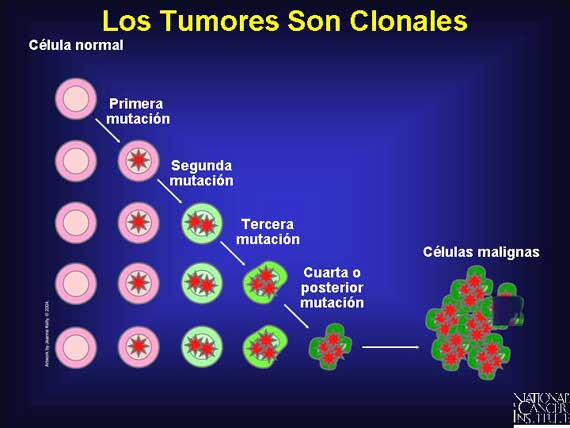
Cada célula, cuando se divide, genera dos células nuevas idénticas. Por lo cual, cuando una célula adquiere una mutación, transfiere esa mutación a su progenie durante el crecimiento y la división celulares. Debido a que las células con mutaciones ligadas al cáncer tienen la tendencia a proliferar más que las células normales, los candidatos celulares para las mutaciones adicionales crecen en número. Las mutaciones continúan acumulándose y son copiadas a las células descendientes. Si una célula finalmente adquiere suficientes mutaciones para convertirse en cancerosa, se derivarán de esa célula transformada individual, subsecuentes células cancerosas. Por lo tanto, todos los tumores son clonales, lo que significa que ellos se originan de una célula madre individual, ya sea que esa primera célula mutante fuera de origen de línea germinal o de origen somático.
|
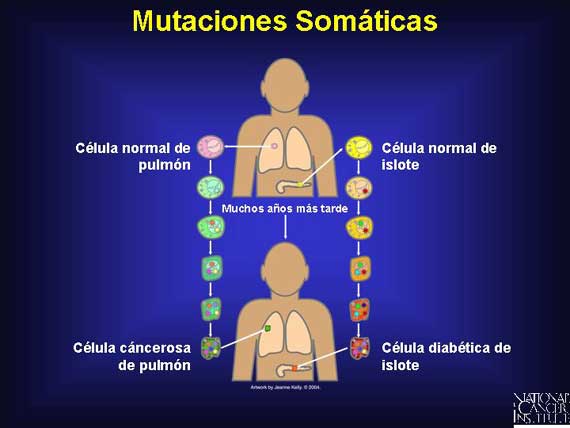
La mayoría de los cánceres humanos resultan de una acumulación de mutaciones somáticas. Las mutaciones somáticas no son transferidas a la siguiente generación. Un periodo de vida de 80 años libre de cáncer no es un logro pequeño. Requiere que tantas como 10 millones de billones de células del cuerpo se copien a sí mismas correctamente. Es fácil ver cómo los errores al azar pueden ocurrir. Estos cambios son adquiridos durante el periodo de vida de una persona proviniendo de exposiciones a carcinógenos y a otros mutágenos o proviniendo de errores al azar no reparados que ocurren durante el crecimiento y la división celulares de rutina. Ocasionalmente, una de estas mutaciones somáticas altera la función de algunos genes importantes, proporcionando una ventaja de crecimiento a la célula en la cual la mutación ha ocurrido. Una clona entonces se origina de esa célula individual.
|
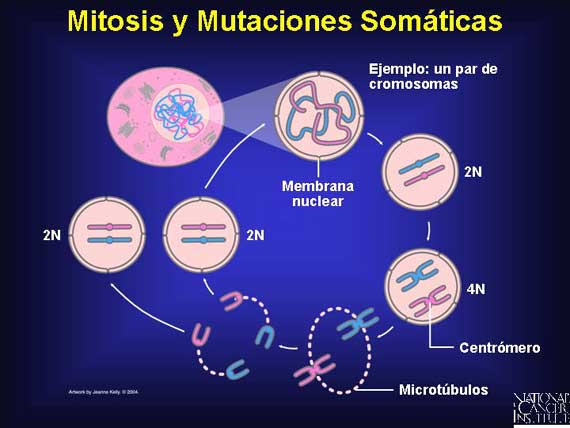
Las células humanas normales con un núcleo que tiene 23 pares de cromosomas se conocen como diploides o 2N para indicar estos pares homólogos. Durante el ciclo de crecimiento celular para las células del cuerpo (somáticas), el ADN de todos los 23 pares-46 cromosomas-se copia a sí mismo (4N). Cuando la célula a continuación se divide mediante un proceso conocido como mitosis, cada célula hija termina con 23 pares ó 2N, un juego completo de cromosomas. Si ocurre una mutación durante el proceso de mitosis, sólo la progenie de la célula somática mutada tendrá la alteración presente.
|
A diferencia de otras células del cuerpo humano, las células germinales que están madurando, como el óvulo o el espermatozoide, deben reducir su número de cromosomas de 46 a 23, de 2N a N. Ellas realizan esto a través de dos divisiones de células especializadas en un proceso conocido como meiosis. Después de que la meiosis se haya completado, cada célula germinal tiene sólo una mitad de los 44 cromosomas originales (o autosomas) del cuerpo además de un cromosoma sexual X ó uno Y.
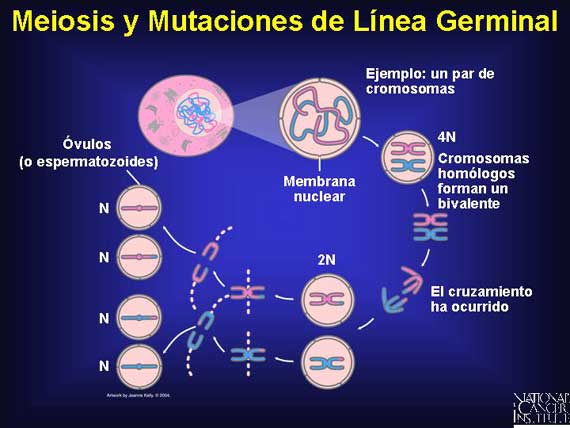
|
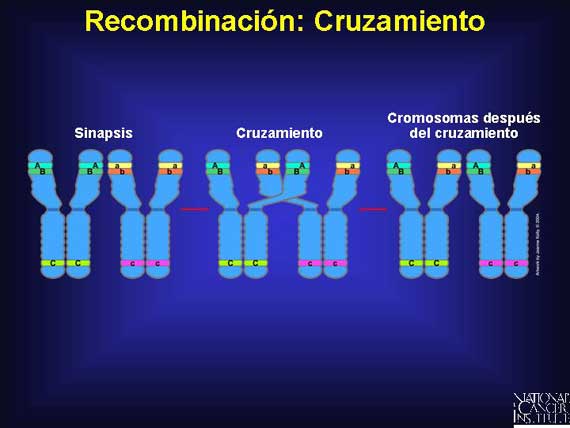
En una etapa temprana de la meiosis, cada par de cromosomas se copia a sí mismo. Estos homólogos se encuentran todos unidos en el centrómero y están enrollados muy apretados alrededor el uno del otro. Inmediatamente antes de que los juegos de duplicados de homólogos se separen y se movilicen hacia un extremo diferente de la célula para completar la primera división, la recombinación puede ocurrir, a medida que el material genético entrelazado se separa. Entonces, más tarde en la meiosis, una segunda división ocurre y hasta los cromosomas dentro de un homólogo se movilizan aparte, dejando sólo un número haploide (n) en cada óvulo o espermatozoide. Si las mutaciones ocurren durante la meiosis, ya sea en el óvulo o en el espermatozoide, éstas serán mutaciones de línea germinal.
Si el óvulo o espermatozoide mutado entonces continúa hacia la fertilización, sus mutaciones de línea germinal serán transferidas a cada célula somática en el nuevo individuo.
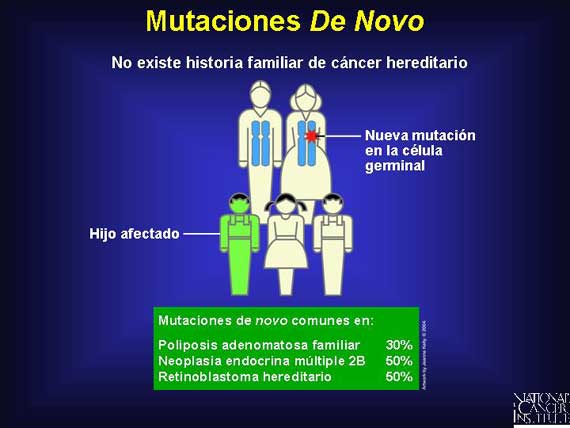
Las mutaciones heredadas tuvieron que iniciar en alguna otra parte y esa otra parte es una mutación de novo. Una mutación de novo es una nueva mutación que ocurre en una célula germinal y entonces se transfiere a uno de la progenie. Todas las mutaciones de línea germinal empezaron como una mutación de novo en algún ancestro. Las mutaciones de novo son comunes en unos cuantos síndromes de susceptibilidad al cáncer heredado.
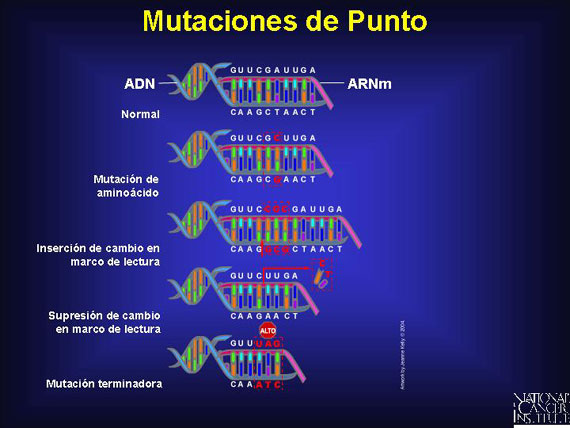
Las mutaciones de punto, cambios de base individual en las secuencias del ADN, son el tipo más común de alteración en el ADN. Ellas pueden tener efectos variables en la proteína resultante.
Una mutación de punto de aminoácidos (missense point mutation, en inglés) substituye un nucleótido por uno diferente, pero deja el resto del código intacto. El impacto de estas mutaciones de punto depende del aminoácido específico que es cambiado y la secuencia de proteína que resulta. Si el cambio es crítico al sitio catalítico de la proteína o a su pliegue o doblado, el daño puede ser severo.
Las mutaciones terminadoras (nonsense mutations, en inglés) son mutaciones de punto que cambian un codón de aminoácido a uno de tres codones de detención, lo cual resulta en la terminación prematura de la proteína. Las mutaciones terminadoras pueden ser causadas por substituciones de pares de bases individuales o por mutaciones de cambio en marco de lectura.
|
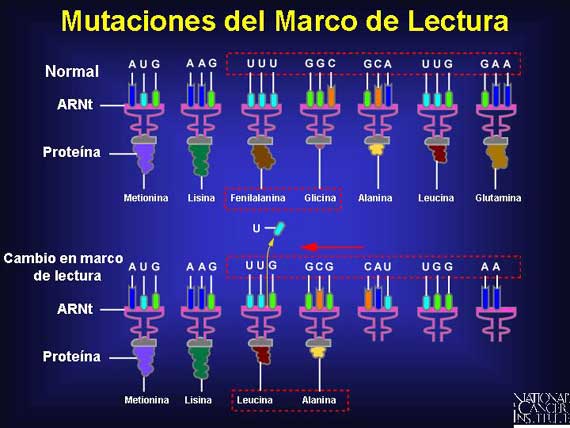
Otro tipo de mutación que puede ocurrir es una mutación de cambio en el marco
de lectura (frameshift mutation). Cuando un gen se copia, la acción
empieza en el núcleo. Ahí una cadena de ARNm copia a la cadena de ADN
exactamente. Codifica para una proteína precisamente, no dejando ningunos
huecos o espacios separando a los tripletes. Este juego de tripletes conectado
se conoce como el marco de lectura (reading frame). Una mutación de cambio en
el marco de lectura es causada por la adición o pérdida de un nucleótido o
nucleótidos. Esto altera el contenido de cada codón triplete que sigue en un
marco de lectura. Las mutaciones de cambio en el marco de lectura por lo
general resultan en una proteína anormal acortada o no funcional y ellas pueden
crear un codón de DETENCIÓN prematuro más adelante. Si el número de pares de
bases añadidas o perdidas es un múltiplo de tres, la proteína resultante puede
ser dramáticamente alterada y su función dependerá de la extensión de estas
alteraciones.
|
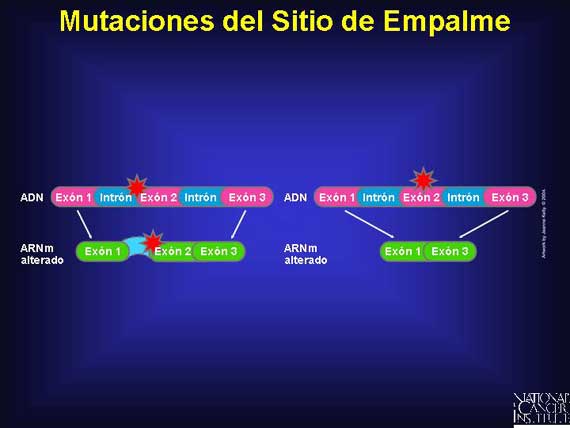
Las mutaciones del sitio de empalme ocurren dentro de los genes en las regiones no codificadas (intrones) exactamente al lado de las regiones codificadas (exones). Ellas pueden tener efectos profundos sobre la proteína resultante, los cuales pueden conducir a la enfermedad. Antes de que el ARNm salga del núcleo, los intrones se remueven y los exones se enlazan juntos. Este proceso se conoce como empalmado. El empalmado es controlado por secuencias específicas de intrones, conocidas como secuencias de empalme-donador y empalme-aceptador, las cuales están al costado de los exones. Las mutaciones en estas secuencias pueden conducir a la retención de segmentos grandes de ADN intrónico por el ARNm, o a exones completos que están siendo empalmados fuera del ARNm. Estos cambios podrían resultar en la producción de una proteína no funcional.
|
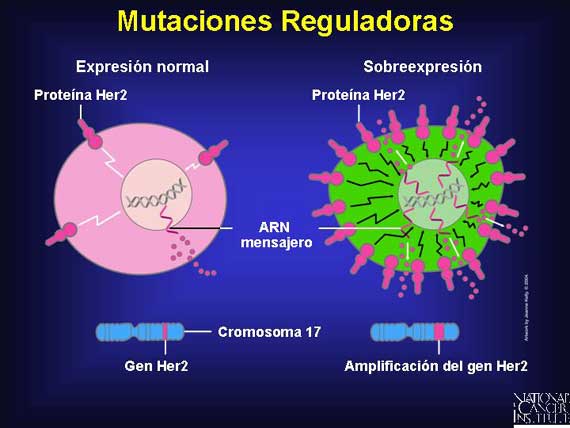
Aunque las mutaciones en la región no codificada son por lo general silenciosas, ese no es siempre el caso. Algunas de las regiones reguladoras más importantes se encuentran en la región no codificada 5' a los costados del gen. Las secuencias promotoras que regulan el gen están localizadas ahí. También, las secuencias amplificadoras que regulan la tasa de actividad de los genes se encuentran en las regiones no codificadas, una distancia considerable desde el gen. Y las regiones represoras de gen, las cuales regulan negativamente la actividad de genes, también existen. Las mutaciones en cualquiera de estas regiones pueden cambiar la tasa de producción de proteínas.
La expresión de la proteína Her2 es un buen ejemplo de la manera en que la amplificación de genes puede tener un impacto regulador sobre el crecimiento de un tumor. En el cáncer del seno, la sobreexpresión de la proteína Her2 resulta de la amplificación de genes en el cromosoma 17. Este aumento en la producción de moléculas señaladoras de crecimiento acelera la tasa del progreso del cáncer.
|
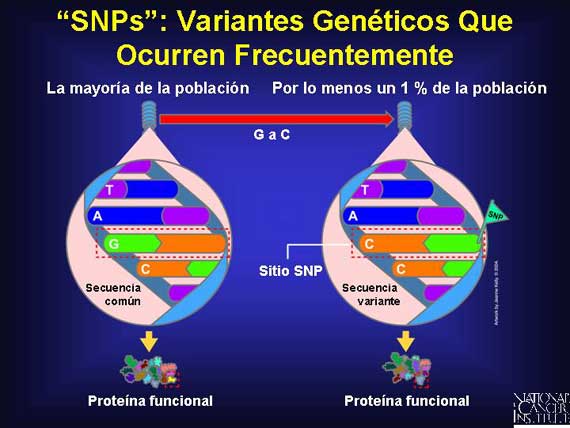
Hay más de un millón de polimorfismos de nucleótido único (single nucleotide polymorphisms o SNPs, en inglés) en el genoma humano. Los SNPs son sitios específicos dentro del genoma humano en los cuales algunas personas tendrán un nucleótido presente mientras que otras personas tendrán uno diferente. Los SNPs empiezan su existencia como mutaciones de punto y ellos eventualmente se establecen en una población. Esta substitución debe ocurrir en una proporción significante (más de un 1 por ciento) de una población grande para ser llamado un SNP. Aquí se presenta un ejemplo: En la secuenca de ADN, TAGC, un SNP ocurre cuando la base G cambia a una base C y la secuencia se convierte en TACC. Cuando los SNPs ocurren dentro de un gen, la proteína que resulta por lo general permanece algo funcional.
|
Las supresiones o inserciones grandes en un cromosoma también pueden conducir al cáncer. Esto puede ocurrir durante la mitosis o durante la recombinación en la meiosis. Las translocaciones ocurren cuando los segmentos de un cromosoma se desprenden y se fusionan a un cromosoma diferente, sin pérdida alguna de material genético. Se ha encontrado que muchas de éstas permiten el desarrollo de tumores. Las inversiones son mutaciones que se originan cuando dos rupturas ocurren en un cromosoma y la pieza se reinserta en orden inverso. Otras anormalidades cromosomales incluyen la no disyunción, el fracaso de los homólogos (pares de cromosomas) para separarse a medida que las nuevas células se dividen.
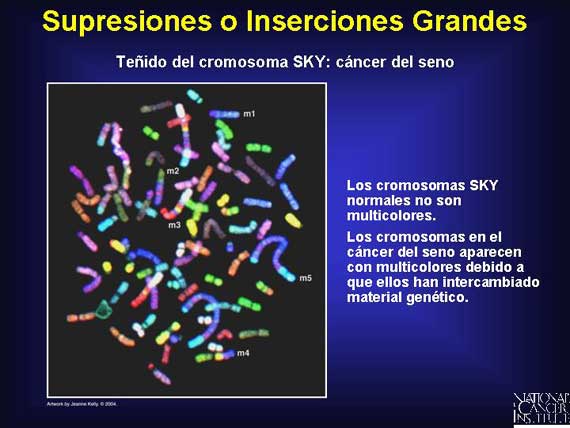
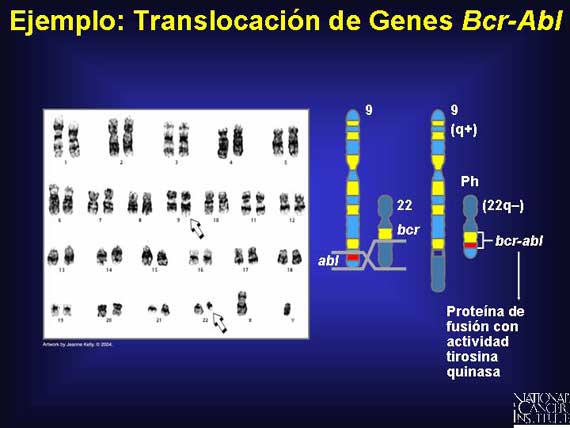
|
En la leucemia mieloide crónica, una translocación ocurre entre los cromosomas 9 y 22. Este rearreglo del material genómico crea un gen de fusión llamado Bcr-Abl que produce una proteína (tirosina quinasa) que se piensa que promueve el desarrollo de la leucemia. El fármaco (medicamento) Gleevec bloquea la activación de la proteína Bcr-Abl.
|
Las mutaciones asociadas con el cáncer, ya sean somáticas o de línea germinal,
ya sean mutaciones de punto o supresiones grandes, alteran a proteínas clave y
a sus funciones en el biosistema humano. Una gran variedad de mutaciones
parecen estar involucradas. Aún las mutaciones en regiones no codificadas, como
por ejemplo en regiones promotoras, amplificadoras o regiones reguladoras
negativas, pueden resultar en subexpresión o sobreexpresión de proteínas
necesarias para la normalidad. Otras mutaciones pueden causar que la producción
de proteínas importantes de control (checkpoint) funcione incorrectamente.
Colectivamente, estas mutaciones conspiran para cambiar a un genoma de normal a
canceroso.
|
El cáncer puede empezar como un nuevo genotipo, es decir, como un cambio en la composición genética de una persona, pero finalmente produce también un nuevo fenotipo. Un fenotipo es la manifestación física de un genotipo en la forma de un rasgo distintivo o enfermedad. El cáncer es conocido por sus genotipos y fenotipos constantemente cambiantes.
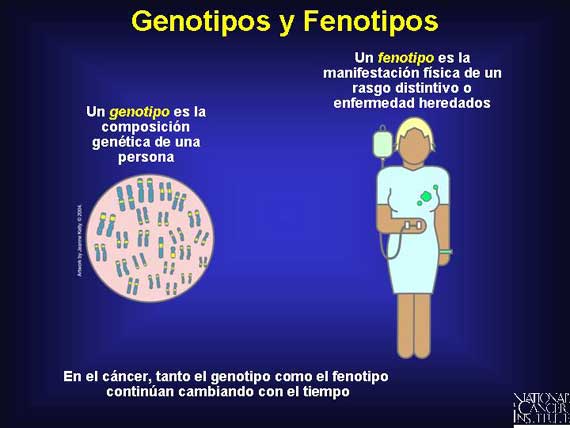
Todos los genotipos no son creados igualmente con respecto a su influencia sobre el fenotipo. Los genes vienen en muchas variedades conocidas como alelos y algunos son más dominantes que otros. En un par de alelos, el efecto de un alelo dominante prevalece sobre el efecto de un alelo recesivo. Y los efectos de un alelo recesivo se vuelven aparentes sólo si el alelo dominante es inactivado o se pierde.
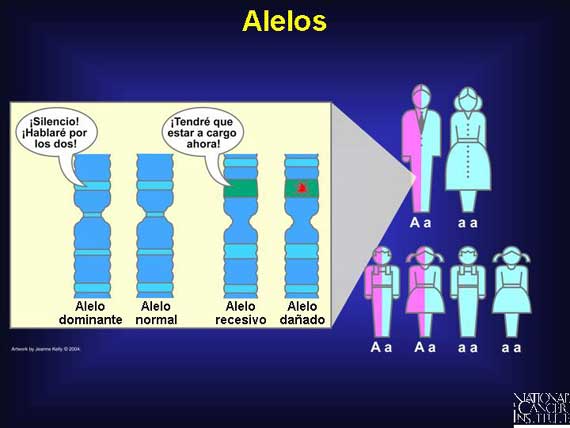
Diferentes mutaciones en el mismo gen pueden resultar en diferentes fenotipos.
Un buen ejemplo es el protooncogen RET. Las mutaciones de línea germinal de RET
conducen a neoplasia endocrina múltiple (multiple endocrine neoplasia o MEN, en
inglés) tipo 2. La enfermedad producida varía dependiendo de en cuál parte del
gen RET la mutación de línea germinal se localiza, por lo que el fenotipo puede
ser MEN-2A, MEN-2B o cáncer medular familiar de la tiroides.

|
Muchos síndromes de susceptibilidad al cáncer son genéticamente heterogéneos
(una mezcla), lo que significa que diferentes mutaciones (genotipos) se pueden
expresar como el mismo fenotipo (p.ej., cáncer). Estas mutaciones diferentes se
pueden localizar dentro del mismo gen pero en diferentes localizaciones
(heterogeneidad de locus) o en diferentes genes por completo (heterogeneidad
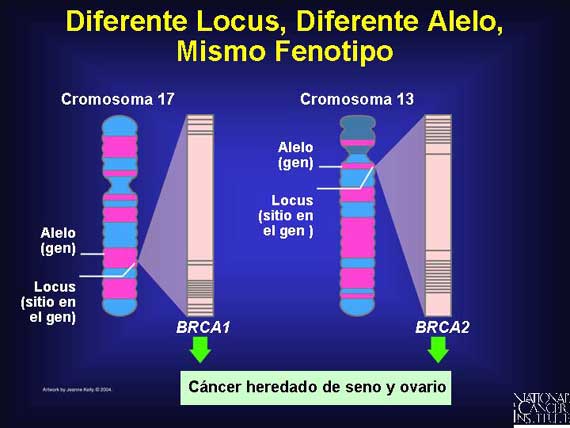
alélica). Por ejemplo, la susceptibilidad al cáncer heredado de seno y de
ovario tiene heterogeneidad tanto de locus como alélica. Más de 500 mutaciones
diferentes han sido identificadas que pueden ocurrir en el gen BRCA1 en el
cromosoma 17 y aumentar el riesgo de una mujer de desarrollar cáncer del seno.
Y más de 300 mutaciones dispersas por todo el gen BRCA2 en el cromosoma 13
están asociadas con la susceptibilidad al cáncer heredado de seno y de ovario.
|
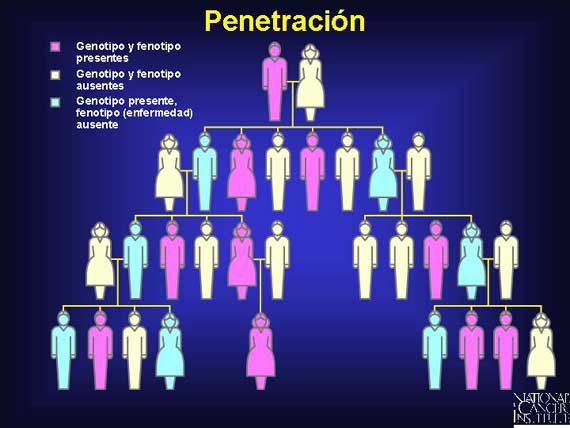
Algunas veces una persona con un alelo dominante expresará un rasgo distintivo,
sin embargo ese mismo genotipo en otra persona permanecerá silencioso. Éste es
un ejemplo de diferencias en penetración. En la genética clásica Mendeliana, si
una persona porta un alelo dominante, el rasgo distintivo será expresado
(genotipo = fenotipo). Sin embargo, si todos los portadores de un cierto alelo
dominante en una población no expresan el rasgo distintivo (mismos
genotipos/diferentes fenotipos), se dice que el gen tiene penetración
incompleta.
|
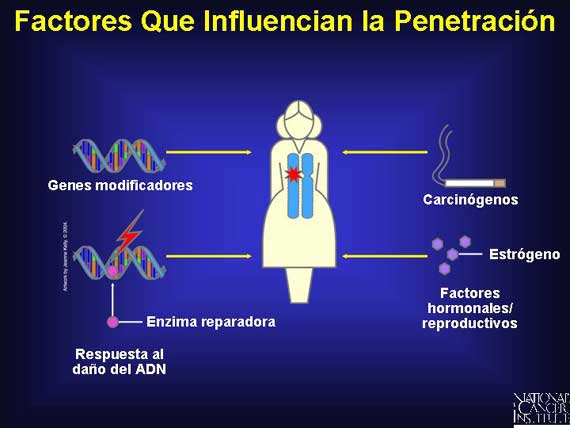
Los genes modificadores afectan la expresión de algunos alelos, lo cual puede aumentar o disminuir la penetración de una mutación de línea germinal, como por ejemplo, un alelo alterado de susceptibilidad al cáncer. La penetración también puede ser afectada por mutaciones en los genes de respuesta al daño del ADN, cuya función normal es la de reconocer y reparar el daño genético. Si la reparación no funciona, las mutaciones se pueden acumular en otros genes, aumentando la probabilidad de que una célula en particular progresará al cáncer.
|
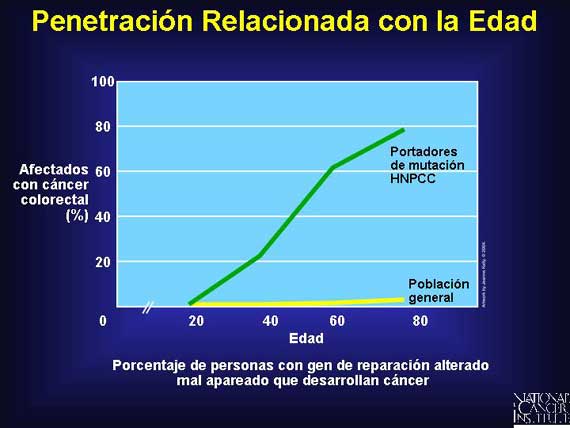
La penetración por lo general está relacionada con la edad, lo que significa que
el rasgo distintivo no es expresado en la mayoría de los portadores al
nacimiento pero ocurre con frecuencia incrementada a medida que los portadores
envejecen. Por ejemplo, las mutaciones de línea germinal en los genes de
reparación mal apareados asociados con el cáncer colorectal no poliposo
hereditario (hereditary nonpolyposis colorectal cancer o HNPCC, en inglés) son
incompletamente penetrantes. Por lo que no todos los individuos que portan
estas mutaciones desarrollarán cáncer colorectal, pero el riesgo aumenta a
medida que las personas envejecen. Alrededor de un 20 por ciento de los
portadores nunca desarrollarán cáncer colorectal.
|
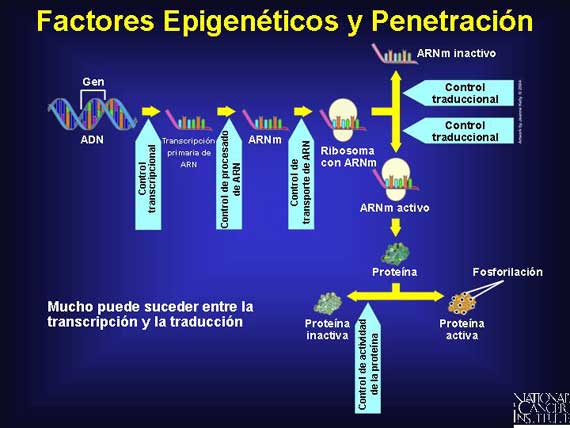
Los factores epigenéticos son mecanismos fuera del gen, como por ejemplo, la exposición de una célula a carcinógenos u hormonas o variaciones genéticas que modifican a un gen o a su proteína por metilación, demetilación, fosforilación o desfosforilación. Estos factores pueden alterar lo que es finalmente expresado; ellos pueden cambiar un fenotipo. Por ejemplo, los factores hormonales y reproductores pueden influir la penetración de ciertas mutaciones ligadas al cáncer. Los cánceres del seno y ovario tienen una probabilidad mayor de ocurrir en mujeres con menarca temprana, menopausia tardía y en aquéllas que tienen su primer bebé después de los 30 años (o que no tienen hijos del todo). Se piensa que estos factores están ligados a la exposición de una mujer a los estrógenos y la progesterona y a sus efectos sobre la diferenciación celular en el seno que ocurren durante el embarazo.
En el cáncer tanto el genotipo como el fenotipo cambian con el tiempo. Los factores epigenéticos juegan un papel muy importante en estos cambios.
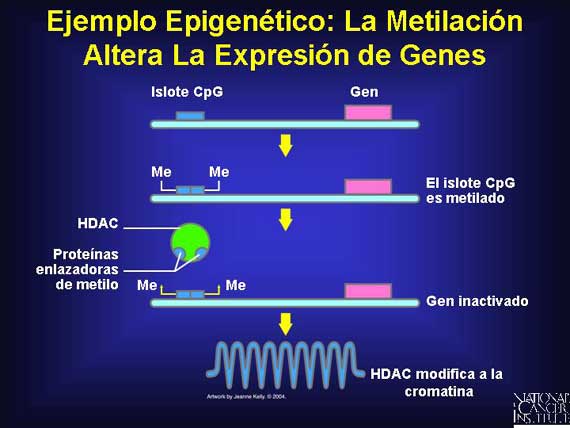
La metilación del genoma puede hacer que áreas se conviertan en
silenciosas. Hay dos tipos de metilación que ocurren. La
metilación de mantenimiento añade grupos metilo a cadenas de ADN
recientemente sintetizadas en puntos opuestos a los sitios metilados en la
cadena madre. Esta actividad asegura que las moléculas hijas de ADN
mantengan un patrón de metilación después de la
división celular. También existe la metilación de novo, la
cual puede añadir grupos de metilo en posiciones totalmente nuevas y
cambiar el patrón en una región localizada del genoma.
Los genes que se deben expresar en todos los tejidos tienen regiones no
metiladas, conocidas como islotes CpG, localizadas en la dirección
opuesta. Por otra parte, los genes que se deben desactivar en los tejidos
diferenciados tienen estos islotes metilados. Esto permite que un complejo
histona-deacetilasa (histone deacetylase complex o HDAC, en inglés) se
una y comprima la forma del material genómico e inactive el gen.
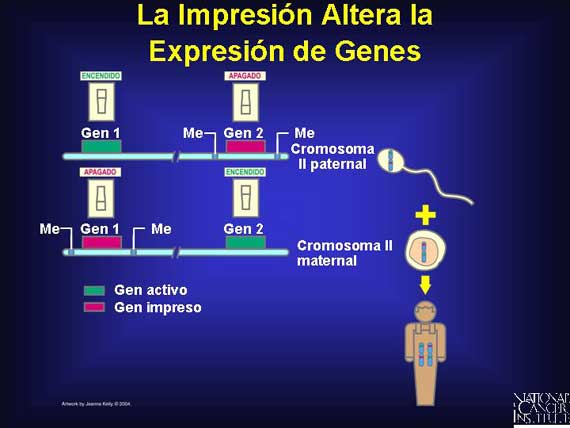
La impresión genómica es un evento poco común en los genomas humanos que ocurre cuando sólo uno de un par de genes presentes en los cromosomas homólogos es expresado debido a que el otro ha sido silenciado por la metilación. Treinta genes en los seres humanos exhiben dicha impresión. Curiosamente, para genes específicos, la copia maternal es la que se escoge para ser silenciada; para otros, la copia paternal es seleccionada.
La frecuencia de portador describe la prevalencia en una población determinada de las mutaciones de línea germinal en un gen específico. Un portador de mutación algunas veces se conoce como heterocigoto debido a que dos alelos diferentes están presentes en un locus dado-uno con una mutación de línea germinal y un alelo normal. En esta transparencia, 2 de cada 10 individuos portan un alelo mutado en un locus de gen particular, de tal manera que la frecuencia de portador es un 20 por ciento.

Algunas poblaciones tienen una prevalencia mayor de alelos específicos asociados con el cáncer que otras poblaciones. Esto puede resultar de un efecto de fundador, lo cual ocurre cuando una población experimenta una contracción rápida y después una expansión en un ámbito aislado. En una población que está geográfica o reproductivamente aislada, un individuo conocido como un fundador porta o desarrolla una mutación de línea germinal que es rara en la población general.
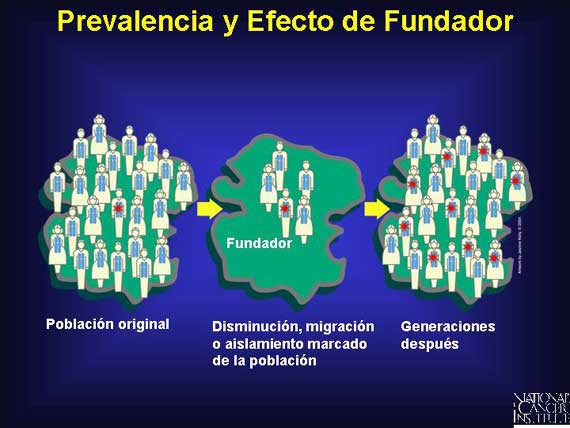
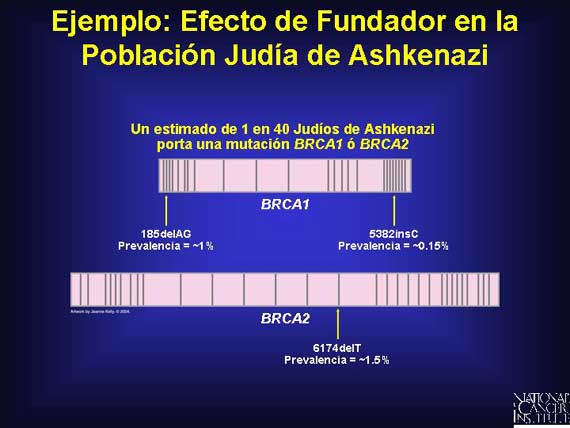
Debido al aislamiento reproductivo, las generaciones posteriores de una población aislada tendrán una frecuencia mayor de una mutación que la población original. Por ejemplo, los judíos de Ashkenazi fueron segregados del resto de la población y vivieron en comunidades separadas por cientos de años. Hoy, un uno por ciento de la población judía de Ashkenazi-una persona en 40-porta una mutación 185delAG en el BRCA1, lo cual los coloca en un riesgo mayor que el riego promedio de desarrollar cáncer del seno y del ovario.
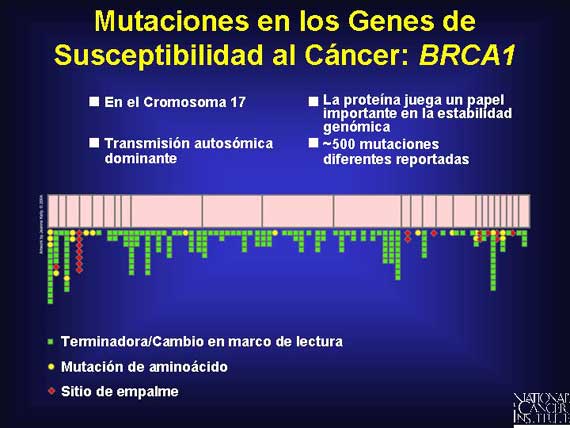
Aquí se presenta un ejemplo de las mutaciones vistas en el gen de susceptibilidad al cáncer del seno BRCA1.
Los individuos que heredan estas mutaciones de línea germinal predispuestas al cáncer portan sus alelos mutados en cada célula de sus cuerpos.
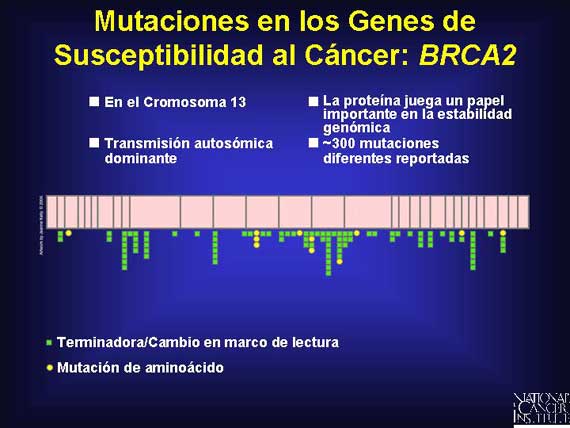
Aquí se presenta un ejemplo de las mutaciones vistas en el gen de susceptibilidad al cáncer del seno BRCA2.
El heredar estos alelos mutados aumenta grandemente el riesgo de una persona de desarrollar cáncer durante su vida. Esto puede explicar por qué los cánceres ligados a las mutaciones de línea germinal en los genes de susceptibilidad frecuentemente ocurren en una edad temprana y en sitios múltiples.
La mayoría de los síndromes hereditarios del cáncer son heredados de manera autosómica dominante.
La herencia dominante ocurre cuando sólo una copia de un alelo es requerida para que un rasgo en particular sea expresado (fenotipo). En la herencia autosómica dominante, las generaciones múltiples expresan los rasgos distintivos, sin saltarse generaciones (asumiendo penetración completa).
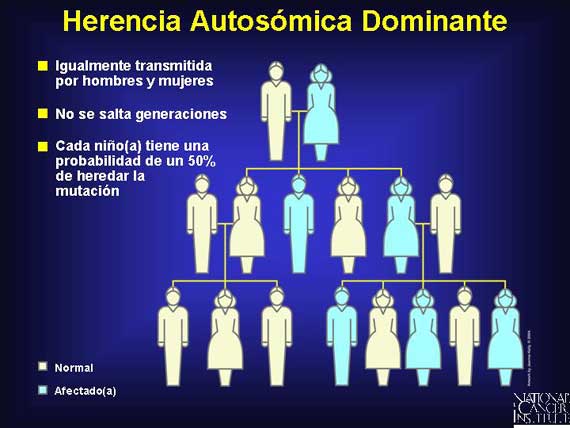
Los síndromes hereditarios del cáncer son relativamente poco comunes, representando solamente alrededor de un 5 a un 10 por ciento de todos los cánceres. Sin embargo, tantos como 50,000 cánceres nuevamente diagnosticados en los EE.UU. cada año están asociados con un síndrome hereditario.

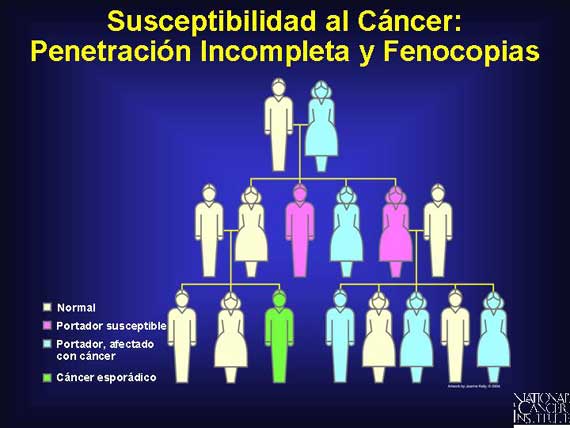
Los individuos que heredan las mutaciones de susceptibilidad al cáncer heredan una predisposición al cáncer, no el cáncer mismo. Algunos portadores de las mutaciones heredan sus genotipos predispuestos de una manera autosómica dominate, sin embargo ellos no desarrollan cáncer, indicando que sus genes alterados son penetrantes incompletamente. Una mutación somática en un segundo alelo es requerida para que el cáncer se desarrolle.
Confundiendo adicionalmente la situación se encuentra el hecho de que las formas esporádicas de cáncer también pueden ocurrir en familias junto con un síndrome hereditario de cáncer. Estos casos de cáncer esporádico se conocen como fenocopias debido a que su fenotipo es similar a aquél de los portadores afectados con mutación, pero su genotipo es diferente. La prueba genética puede determinar si el cáncer es hereditario o de naturaleza esporádica.
|
En este linaje de una familia con una mutación BRCA1, los números debajo de cada persona indican la edad en que se diagnosticó el cáncer por primera vez, si estaba afectada; edad al morir, si falleció; y edad al tiempo de la entrevista, si está viva. Esta familia ejemplifica varios contrastes del cáncer hereditario de seno y de ovario. El hecho de que la mutación es transferida por transmisión autosómica dominante es evidente en el sentido de que aproximadamente un 50 por ciento de los miembros de la familia en cada generación portan la mutación. Observe que un padre no afectado transmite la mutación a su hija afectada, mostrando que la transmisión de la mutación BRCA1 puede ocurrir a través de cualquiera de los padres. Observe la alta penetración de la enfermedad y la edad temprana de inicio. La penetración es incompleta, aunque alta, como es mostrada por un portador femenino que vive hasta la edad de 86 años y otra que vive hasta la edad de 92 años sin un diagnóstico de cáncer del seno o del ovario.

|
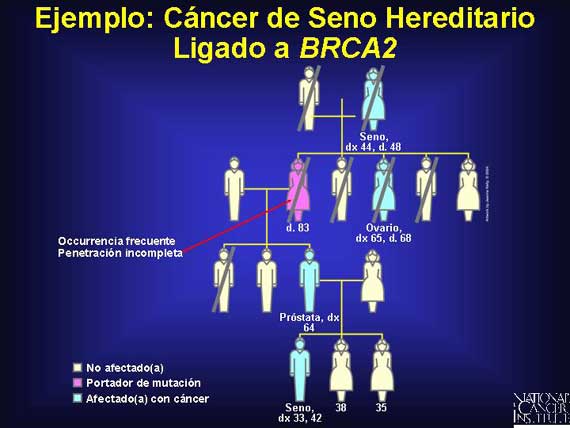
En este linaje, nuevamente se revela que la mayoría de la mutaciones en los
genes de susceptibilidad al cáncer son de línea germinal y se trasmiten como
rasgos dominantes con penetración incompleta. Observe que el portador femenino
vive hasta la edad de 83 años y muere de causas naturales aún cuando su madre
fue afectada por cáncer del seno y murió a la edad de 48 años. También observe
que dos hombres fueron diagnosticados con cáncer: uno del seno y el otro con
cáncer de la próstata. Los hombres que heredan un gen anormal BRCA2 tienen un
riesgo incrementado (80 veces el riesgo por vida de los hombres sin la
mutación) de desarrollar el cáncer del seno masculino. Ellos también tienen una
probabilidad de tres a siete veces mayor que los hombres sin la mutación de
desarrollar cáncer de la próstata.
|
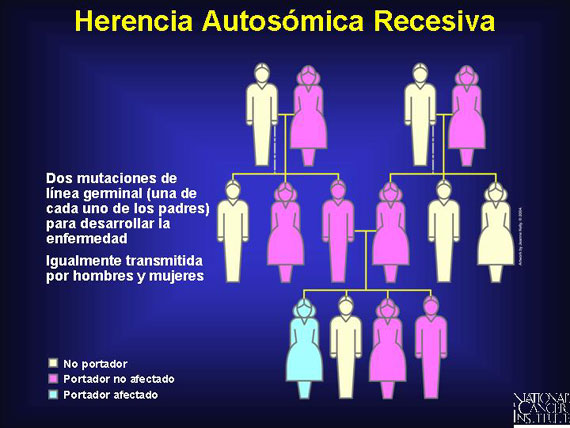
En la herencia autosómica recesiva, dos copias del alelo son requeridas para que se exprese el rasgo distintivo. Los portadores de un alelo de enfermedad no desarrollarán la enfermedad y varias generaciones pueden estar no afectadas, conduciendo a la apariencia de generaciones saltadas. Los hombres y las mujeres son igualmente afectados. Si ambos padres portan una copia del alelo recesivo, uno de cuatro de la progenie, en promedio, expresará el rasgo distintivo.

En la herencia ligada al cromosoma X, el gen de interés se encuentra en el cromosoma X, no en un autosoma. Debido a que las mujeres tienen 2 cromosomas X, ellas deben heredar dos copias del alelo de enfermedad para expresar el fenotipo de la enfermedad. Las mujeres con sólo un alelo mutado son portadoras.
Los hombres son más frecuentemente afectados debido a que ellos sólo tienen un cromosoma X y necesitan sólo un alelo mutado para expresar un fenotipo de la enfermedad. Todos los hombres que heredan una copia del cromosoma X anormal son afectados por la enfermedad (asumiendo un 100 por ciento de penetración).
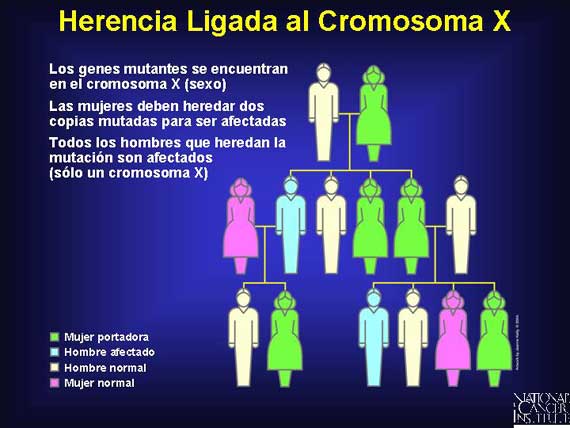
|
La susceptibilidad hereditaria al cáncer del seno ocurre en varias otras
condiciones genéticas raras. El cáncer del seno es la manifestación adulta más
común del síndrome de Li-Fraumeni, un síndrome de cáncer múltiple causado por
mutaciones de línea germinal en el gen TP53. El cáncer del seno también es la
malignidad más frecuentemente diagnosticada en el síndrome de Cowden, una
condición con mutaciones de línea germinal en el gen PTEN. Tanto los tumores
del seno benignos como malignos ocurren en el síndrome de Muir-Torre, una
condición relacionada con el cáncer de colon no poliposo hereditario (HNPCC,
por sus iniciales en inglés), caracterizada por las mutaciones de línea
germinal en los genes de reparación no apareados de ADN, MSH2 y MLH1. Los
pacientes con síndrome de Peutz-Jeghers presentan pigmentación anormal, pólipos
gastrointestinales y, si son mujeres, ellas se encuentran en riesgo
incrementado de desarrollar cáncer del seno y experimentando enfermedad
bilateral con inicio en etapa temprana.

|
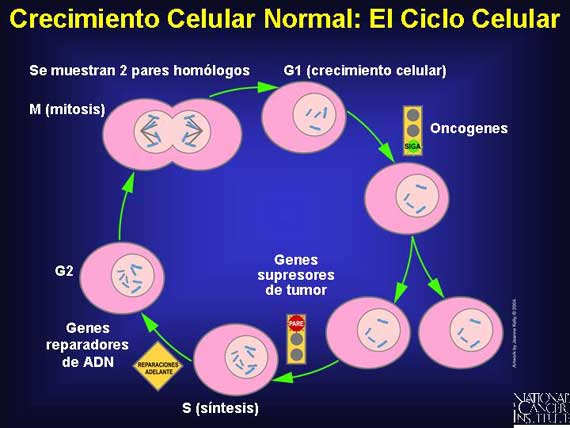
El ciclo celular es un proceso crítico al cual una célula se somete con el fin de copiarse a sí misma exactamente. La mayoría de los cánceres tienen mutaciones en las señales que regulan el ciclo de crecimiento y división de la célula. La división celular normal es requerida para la generación de nuevas células durante el desarrollo y para el reemplazo de las células viejas a medida que ellas mueren.
La mayoría de las células permanecen en interfase, el periodo entre las divisiones celulares, por lo menos un 90 por ciento del ciclo celular. La primera parte de la interfase se conoce como G1 (por el primer intervalo), seguido de la fase S (por la síntesis del ADN), después G2 (por el segundo intervalo). Durante la fase G1, hay crecimiento rápido y actividad metabólica, incluyendo la síntesis del ARN y proteínas. El crecimiento celular continúa durante la fase S y el ADN se replica. En la fase G2, la célula continúa creciendo y se prepara para la división celular. La división celular (mitosis) es referida como la fase M. Las células que no se dividen por periodos largos no replican su ADN y se considera que están en G0.
En células normales, los genes supresores de tumor actúan como señales de frenado durante la fase G1 para detener o hacer más lento el ciclo celular antes de la fase S. Los genes reparadores de ADN son activos durante todo el ciclo celular, particularmente durante la fase G2 después de la replicación del ADN y antes de que los cromosomas se preparen para la mitosis.
|
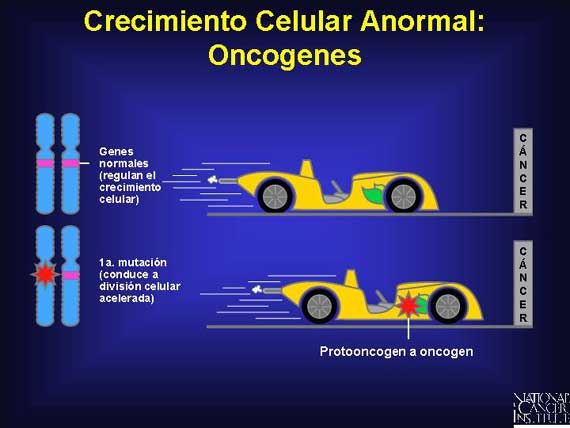
La mayoría de los cánceres tienen mutaciones en los protooncogenes, los genes normales involucrados en la regulación del crecimiento celular controlado. Estos genes codifican a las proteínas que funcionan como factores de crecimiento, receptores de factor de crecimiento, moléculas transmisoras de señales y factores de transcripción nuclear (proteínas que se unen a los genes para iniciar la transcripción). Cuando el protooncogen es mutado o sobreregulado, se conoce como un oncogen y resulta en crecimiento y transformación celulares no reguladas. A nivel celular, sólo una mutación en un alelo individual es suficiente para activar un papel oncogénico en el desarrollo del cáncer. La probabilidad de que dicha mutación ocurrirá aumenta a medida que una persona envejece.
|
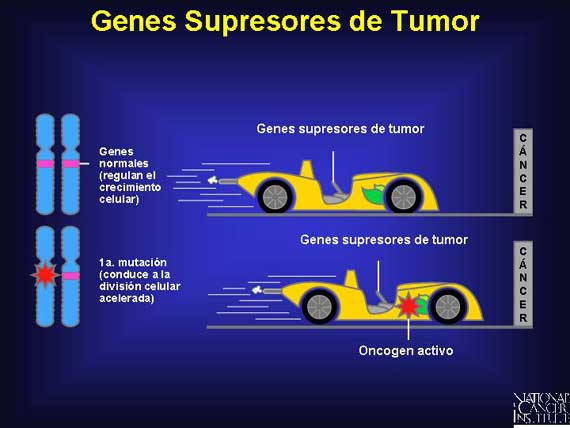
La mayoría de los genes de susceptibilidad al cáncer son genes supresores de
tumor. Los genes supresores de tumor son simplemente un tipo de los muchos
genes que funcionan equivocadamente en el cáncer. Estos genes, bajo
circunstancias normales, suprimen el crecimiento celular. Algunos lo hacen así
codificando a factores de transcripción para otros genes necesarios para
retardar el crecimiento. Por ejemplo, el producto de la proteína del gen
supresor TP53 se conoce como la proteína p53. Se une directamente al ADN y
conduce a la expresión de genes que inhiben el crecimiento celular o activan la
muerte celular. Otros genes supresores de tumor codifican proteínas que ayudan
a controlar el ciclo celular.
|
Ambas copias de un gen supresor de tumor deben perderse o mutar para que el cáncer ocurra. Una persona que porta una mutación de línea germinal en un gen supresor de tumor tiene sólo una copia funcional del gen en todas las células. Para esta persona, la pérdida o mutación de la segunda copia del gen en cualquiera de estas células puede conducir al cáncer.
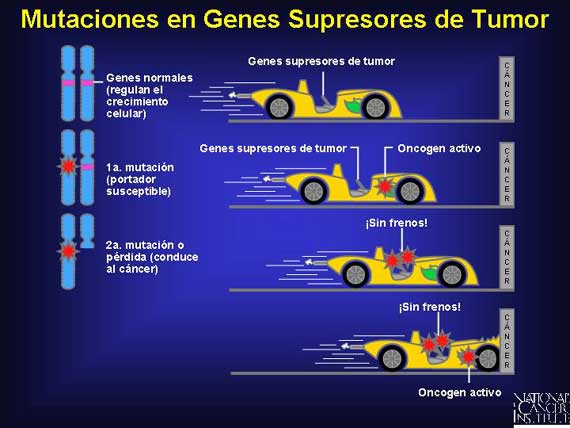
|
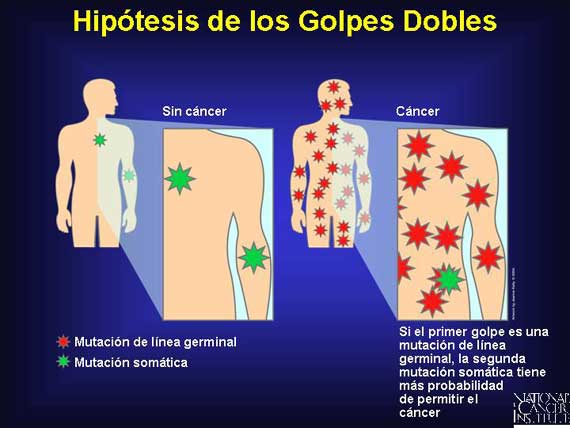
En 1971, el Dr. Alfred Knudson propuso la hipótesis de golpes dobles (two-hit hypothesis, en inglés) para explicar el inicio en una etapa temprana en sitios múltiples del cuerpo de una forma heredada de cáncer conocida como retinoblastoma hereditario, El heredar una copia de línea germinal de un gen dañado presente en cada célula del cuerpo no era suficiente para permitir que este cáncer se desarrollara. Sin embargo, un segundo golpe a (o pérdida de) la copia buena en el par de genes podría ocurrir somáticamente, produciendo cáncer. Esta hipótesis pronosticó que la probabilidad de que un portador de mutación de línea germinal obtuviera una segunda mutación somática en cualquiera de los sitios múltiples en las células de su cuerpo era mucho mayor que la probabilidad de que un no portador obtuviera dos golpes en la misma célula.
Los supresores de tumor actúan recesivamente a nivel fenotípico (ambos alelos deben ser mutados/perdidos para que se desarrolle el cáncer), pero la mutación de línea germinal de "primer golpe" a nivel genotípico es en realidad heredada de un modo autosómico dominante.
|
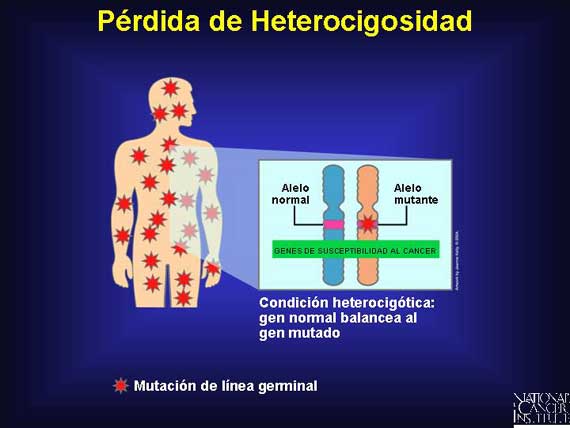
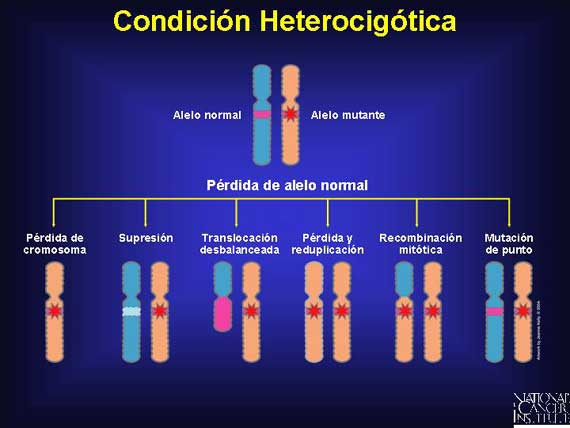
Hay varias maneras en que una célula puede sufrir pérdida de heterocigosidad. Un cromosoma completo que contiene un alelo normal se puede perder debido a una falla de los cromosomas de segregarse apropiadamente durante la mitosis (no disyunción). Alternativamente, un intercambio no balanceado de material genético puede ocurrir en un proceso conocido como translocación, resultando en la pérdida de una región cromosómica conteniendo el gen normal. Algunas veces cuando un gen normal se pierde, una reduplicación del cromosoma restante con un gen anormal ocurre, dejando a la célula con dos copias de genes anormales. Los genes normales también pueden perderse durante los eventos de recombinación mitótica normales o como consecuencia de una mutación de punto en el segundo alelo, conduciendo a la inactivación de la contraparte normal.
|
En los síndromes de cáncer heredado, los individuos son conocidos como
heterocigotos (teniendo uno o más pares de genes disimilares) debido a que
ellos empiezan la vida con una mutación de línea germinal en uno de los alelos
ligados a la susceptibilidad al cáncer, pero es balanceada por una contraparte
normal. Estos individuos están predispuestos al cáncer debido a que todas sus
células ya han sido sometidas al primer golpe en los genes ligados con el
cáncer. Si el gen supresor normal críticamente necesario que balancea esta
mutación de línea germinal se pierde en algún momento durante la vida del
individuo, una condición conocida como pérdida de la heterocigosidad (loss of
heterozygosity o LOH, en inglés) ocurre.
|
Algunas mutaciones ligadas al cáncer parecen involucrar una incapacidad de uno o muchos de los sistemas de reparación de la célula. Un ejemplo de dicho error involucra a la reparación de mal apareamiento del ADN. Después de que el ADN se copia a sí mismo, las proteínas de los genes de reparación de mal apareamiento actúan como correctores de prueba para identificar y corregir los apareamientos incorrectos. Si una pérdida o mutación ocurre en los genes de reparación de mal apareamiento, es muy probable que las mutaciones esporádicas se acumulen. Otros errores en la reparación pueden involucrar el corte incorrecto de bases-o de nucleótidos completos-a medida que las proteínas de reparación intentan reparar el ADN después de que moléculas muy voluminosas, como por ejemplo los carcinógenos en los cigarrillos, se hayan fijado. Esto se conoce como reparación de escisión defectuosa. Algunas veces ambas cadenas de ADN sufren roturas al mismo tiempo y la reparación de recombinación defectuosa ocurre. Cualquiera de estos errores puede permitir que las mutaciones persistan, se copien y eventualmente contribuyan al desarrollo de cáncer.
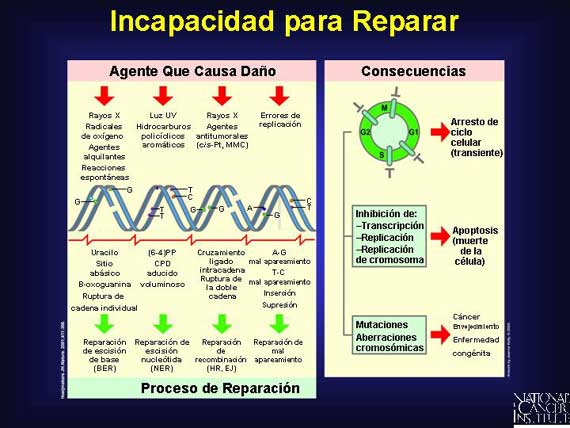
|
Mucha información aún evade a nuestro entendimiento acerca de la
susceptibilidad al cáncer. El cáncer del seno es un buen ejemplo de qué tan
incompleta imagen tenemos.
La mayoría de las mujeres con una historia familiar de cáncer del seno NO
portan mutaciones de línea germinal en los genes individuales de
susceptibilidad al cáncer altamente penetrantes y sin embargo grupos familiares
continúan apareciendo con cada nueva generación.
Alrededor de un 5 a un 10 por ciento de los casos de cáncer del seno están
ligados a las mutaciones de línea germinal en genes individuales de
susceptibilidad al cáncer altamente penetrantes, como por ejemplo, BRCA1 y
BRCA2. La predisposición genética y susceptibilidad al cáncer fuertes en estas
familias se transmiten de una manera autosómica dominante.
Sin embargo, otro 15 a 20 por ciento de cánceres del seno están asociados con
alguna historia familiar pero no hay evidencia de dicha transmisión autosómica
dominante. Estos casos no son muy bien entendidos. Posiblemente algunas
interacciones ambientales o de gen múltiple contribuyen a una muy baja
penetración de los genes de susceptibilidad o posiblemente estén involucradas
mutaciones aún por descubrirse.

|
Muchos aspectos permanecen desconocidos acerca del papel que juegan los factores epigenéticos y el cáncer. Los cambios epigenéticos son modificaciones reversibles a genes o proteínas que ocurren en el tumor y su microambiente. Las moléculas modificadoras epigenéticas han sido observadas realizando cambios tumor-amistosos, no mutacionales, en un biosistema ya confuso. Por ejemplo, al metilar fuertemente a genes o a regiones promotoras, la actividad de los genes que es sumamente importante para contrarrestar el impulso de un tumor hacia la metástasis se desactiva. O los ácidos ribonucléicos no codificados se entrometen de manera epigenética, interfiriendo con la regulación de crecimiento de la célula o con sus intentos de reparar el daño.
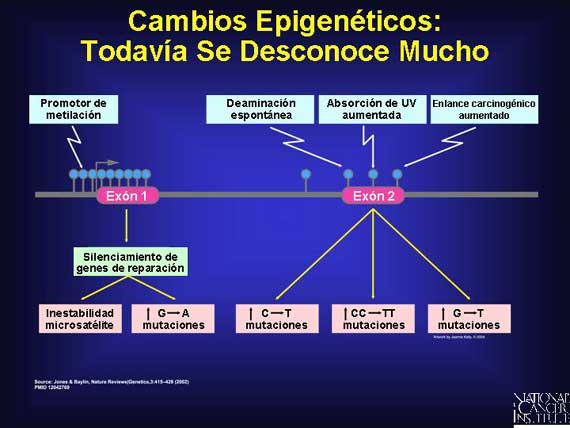
|
Además de los oncogenes y los genes supresores de tumor, la mayoría de los cánceres adquieren varias otras mutaciones clave que permiten que el cáncer progrese. Aunque los investigadores aún no conocen todas las mutaciones involucradas, ellos las han organizado en términos de sus actividades en apoyo del crecimiento del tumor y las metástasis. Además de las contribuciones de oncogenes y genes supresores mutados, las mutaciones genómicas adicionales permiten la invasión de tejidos vecinos, evasión de la detección por el sistema inmunológico, reclutamiento de una nueva provisión de sangre, diseminación y fijación como blancos a nuevos sitios y la penetración y reinvasión a través de la sangre y capas de tejido nuevos. Con el tiempo, la metástasis con éxito ocurre.

|
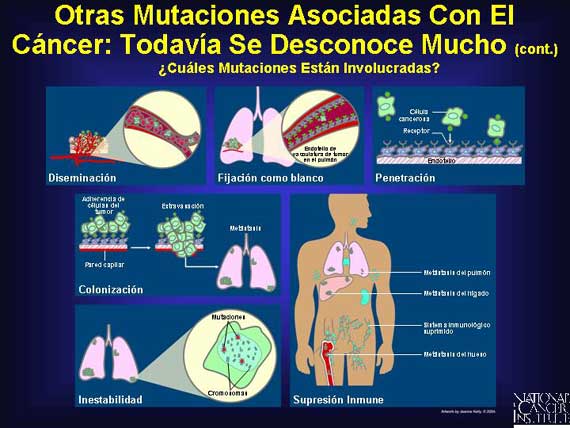
|
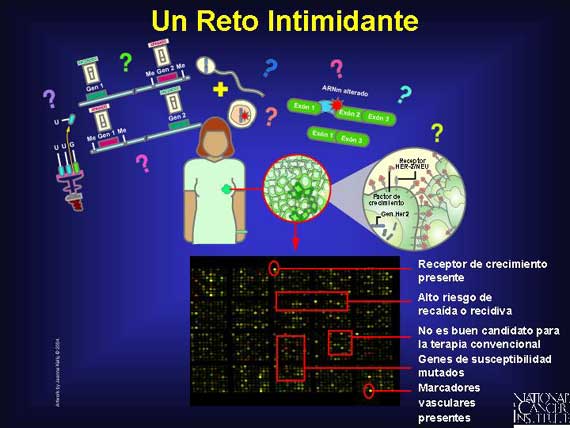
Un análisis completo del genoma del cáncer permanece siendo un reto
intimidante. Actualmente no existe una tecnología individual que detecte todos
los tipos de anormalidad-deleciones, rearreglos, mutaciones de punto,
inserciones de cambio en el marco de lectura, amplificaciones, impresión y
cambios epigenéticos-implicados en el cáncer. Los análisis de microarreglos
(microformaciones) y de chip de genes, sin embargo, están empezando a revelar
algunos drivers (conductores) genómicos clave. (Por favor, consulte la
presentación de Diagnósticos Moleculares para obtener información adicional).
Muchos estudios clínicos ahora incluyen perfiles genómicos de pacientes con
cáncer como indicadores pronósticos y diagnósticos. Los perfiles genómicos son
usados hasta para monitorear dónde y cómo el genoma del cáncer ha sido golpeado
durante las terapias molecularmente destinadas. Examinar y compartir todos
estos datos debe eventualmente ayudar a los oncólogos a integrar mejor los
cambios genotípicos y fenotípicos que ocurren en un biosistema durante el
progreso del cáncer. Este conocimiento será utilizado para lograr
intervenciones mejores y en una etapa más temprana en los pacientes con cáncer.
|