|
| |
||
| |
|||||||||||||||||
|
|||||||||||||||||
|
|||||||||||||||||
|
EID Home | Ahead of Print | Past Issues | EID Search | Contact Us | Announcements | Suggested Citation | Submit Manuscript PDF
Version | Download Adobe
Acrobat | Comments | |
|
Research Epidemic and Nonepidemic Multidrug-Resistant Enterococcus faeciumHelen L. Leavis,*† Rob J.L. Willems,† Janetta Top,† Emile Spalburg,†
Ellen M. Mascini,* Ad C. Fluit,* Andy Hoepelman,* Albert J. de Neeling,†
and Marc J.M. Bonten*
Enterococcus faecium has become an important nosocomial pathogen, especially in immunocompromised patients, creating serious limitations in treatment options because of cumulative resistance to antimicrobial agents (1). In the United States, the emergence of nosocomial E. faecium infections was characterized by increasing resistance to ampicillin in the 1980s and a rapid increase of vancomycin resistance in the next decade (1,2). The emergence of vancomycin-resistant E. faecium (VREF) in the United States illustrates the transmission capacities of bacteria and the possibility of a postantibiotic era for nosocomial infections in critically ill patients. The global epidemiology of VREF is not well understood. In the United States, prevalences of colonization and infection are high among hospitalized patients, but a community reservoir of VREF in healthy persons or animals seems to be absent (3,4). In contrast, in Europe, colonization and infection rates within hospitals remain low, although colonization among healthy persons and animals is prevalent (5–10). Previous studies suggested host-specificity of VREF genogroups (11), and isolates associated with nosocomial outbreaks seemed to be genetically distinct from nonepidemic VREF isolated from humans and animals (12). The differences between epidemic and nonepidemic isolates were based on genetic relatedness, as determined by amplified fragment length polymorphism analysis (AFLP), and the presence of an identical sequence of the purK housekeeping gene in epidemic strains (12). A recently developed multilocus sequence typing scheme for E. faecium confirmed that epidemic isolates belonged to a specific genetic lineage (13). Moreover, a variant of the esp gene, which has been found to be more prevalent among isolates of E. faecalis associated with infections (14), was found in all but one epidemic hospital-derived VREF isolate and not among community-derived VREF (12). Subsequently, other investigators described the variant esp gene in vancomycin-susceptible E. faecium (VSEF), and this gene appeared to be predominantly present among clinical isolates (15–18).These findings suggest the existence of a specific subpopulation of E. faecium, comprising both VREF as well as VSEF, associated with hospital outbreaks and infections. In this study, we further investigated the genetic relationship between VREF and VSEF isolates, derived from different epidemiologic sources, such as hospital outbreaks, infections, and colonization among hospitalized patients and healthy persons. The genetic relatedness was linked to the presence of the variant esp gene and antibiotic resistance to ampicillin and vancomycin. On the basis of our findings, we constructed an evolutionary scheme describing the sequential steps in the development and selection of ampicillin- and vancomycin-resistant E. faecium strains. Materials and MethodsBacterial Strains and Growth ConditionsIsolates of VREF (n=108) were collected from nosocomial epidemics (n=16), clinical infections (n=20), clinical surveys (n=36), and community surveys (n=36) (Table 1). The genotypes of these isolates have been described previously (11,12). Strains were considered epidemic if they were isolated from patients treated in the same hospital, in the same ward and with an overlapping time-relationship, and if AFLP patterns showed at least 90% similarity (12). Epidemic isolates were recovered from clinical sites, blood and urine, as well as from feces. Only one representative isolate from each outbreak was used for analysis. The number of patients involved in each outbreak varied from 4 to >50 (12,19-23). Isolates were considered to be derived from a clinical infection if obtained from a clinical specimen, such as the blood, urine, and wounds. All surveillance isolates, from patients and healthy persons, were isolated from fecal samples. Surveillance isolates were either from the community or from clinical surveillance when obtained from hospitalized patients. The hospital-stay duration of these patients, when cultures were obtained, was not available. Isolates of VSEF (n=92) were derived from clinical infections (n=73), clinical surveys (n=5), and community surveys (n=14). The isolates from clinical infectious sites were obtained from the SENTRY Antimicrobial Surveillance Program, and originated from different hospitals in several European countries (Portugal, Germany, United Kingdom, France, Spain, Italy, Austria, Turkey, Switzerland, Greece, and Poland). Fifty-seven strains were blood isolates, 5 were isolated from urine, 8 from wounds, and 2 from respiratory tract specimens. Patient information was not available. All VSEF isolates derived from clinical surveys of fecal samples were from the University Hospital Maastricht. All VSEF isolates from community surveys of fecal samples were collected in the Netherlands. All bacterial isolates were collected during the 1990s. Identification and Susceptibility TestingEnterococci were identified to the species level and were tested for the presence of the vanA gene by using a multiplex PCR described by Dutka-Malen et al. (24). Vancomycin and ampicillin/amoxicillin susceptibilities were determined by standard agar dilution methods, according to the National Committee for Clinical Laboratory Standards (NCCLS) guidelines (25). We considered MICs >16 µg/mL for ampicillin or amoxicillin and >8 for vancomycin to be resistant. Esp PCRAll strains were screened for esp by PCR, with two different primer sets (esp 11 [5´-TTGCTAATGCTAGTCCACGACC-3´] to esp 12 [5´-GCGTCAACACTTGCATTGCCGAA-3´] and 14F [5´-AGATTTCATCTTTGATTCTTGG-3´] to 12R [5´-AATTGATTCTTTAGCATCTGG-3´]). PCR conditions included an initial denaturation at 95°C for 15 min for activation of the HotStarTaq DNA polymerase (QIAGEN GmbH, Hilden, Germany), followed by 30 cycles of 94°C for 30 sec, 52°C for 30 sec, and 72°C for 1 min, followed by an extension at 72°C for 7 min. Reactions were performed in 25 µL by using the HotStarTaq Master Mix (QIAGEN GmbH). Strains negative in PCR were checked for the presence of the esp gene by Southern hybridization, as described previously (12). For this check, we generated an esp-specific probe (956 bp) using primers esp 11 and esp 12 (previous explanation). Sequencing PurKThe purK gene encodes a phosphoribosylaminoimidazole carboxylase ATPase subunit involved in purine biosynthesis and is one of the seven housekeeping genes selected for multilocus sequence typing of E. faecium (13). A 492-bp fragment of the purK gene of a selection of strains, divided over all genogroups, was sequenced by using primers 5´-GCAGATTGGCACATTGAAAGT-3´ and 5´-TACATAAATCCCGCCTGTTTC/T-3´. PCR conditions included an initial denaturation at 95°C for 3 min, followed by 35 cycles of 94°C for 30 sec, 50°C for 30 sec, and 72°C for 30 sec, followed by an extension at 72°C for 5 min. Reactions were performed in 50 µL by using buffers and Taq polymerase (SphaeroQ, Leiden, Netherlands). The PCR products were purified with a PCR purification kit (QIAGEN GmbH) according to the manufacturer’s instructions. Subsequently, purified PCR products were sequenced directly with the ABI PRISM Big Dye Terminators cycle sequencing kit on an ABI PRISM DNA analyzer (Applied Biosystems, Foster City, CA). Sequences were aligned with BioNumerics (v. 2.5, Applied Maths, Kortrijk, Belgium) software. AFLPAFLP typing and computer analysis of AFLP-generated patterns of VSEF was done as described previously (11) with minor modifications. Briefly, chromosomal DNA was digested with CfoI and EcoRI and ligated to a single adapter with CfoI and EcoRI protruding ends in a simultaneous reaction, followed by PCR using adapter-specific primers. The amplification products were separated and detected by using POP6-polymer on an ABI PRISM 3700 DNA Analyzer (Applied Biosystems). For each sample, 1 µL of the PCR reaction mixture (8 x diluted) was added to 9 µL of Hi-Di Formamid containing 12.5 µL/mL of the internal size marker (GeneScan-500–labeled with the red fluorescent dye 6-carboxy-x-rhodamine) in a MicroAmp Optical 96-well reaction plate (Applied Biosystems). The analyses were run in 3 hours. Genescan software (Applied Biosystems) was used for collection of data during the analysis and the data were subsequently exported into BioNumerics (Applied Maths) for further analysis. The Pearson product moment correlation coefficient was calculated, and the unweighted pair group method with arithmetic averages was used for cluster analysis. Using this methodology, we described four genogroups of VREF (11). We analyzed all isolates of VSEF and defined a cluster of isolates as a set of individual strains with AFLP patterns that shared at least 65% of the banding patterns (criterion defined for four genogroups [11]). Subsequently, to determine the matching genogroup, we compared AFLP banding patterns of each individual VSEF isolate with AFLP banding patterns of a library of 404 VREF, representing the four different AFLP genogroups. The library included VREF recovered from pigs (n=108) and nonhospitalized persons (n=28) as representatives of genogroup A, and strains from poultry (n=32), hospitalized patients (n=196), and calves (n=40), representing genogroups B, C, and D, respectively (11,12). VSEF isolates were identified by using the identification module in BioNumerics. The genetic distance used for further analysis is 100 minus the calculated Pearson product moment correlation similarity coefficient. The degree of matching was expressed by an identification factor, which is the quotient of the average genetic distance between the tested strain and each of the isolates in the genogroup divided by the average genetic distance within a genogroup. If the average distance of the tested strain to each of the genogroup members is almost equal to the average distance among all strains in a genogroup, the identification factor will approach the value of one. So the lower the value of the identification factor, the more likely the test strain belongs to a particular genogroup. Results
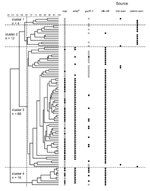
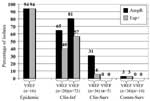
Using our predefined cutoff points for cluster analysis, we identified four clusters of VSEF (Figure 1). VSEF clustering seemed to be source-related. Clusters 1 (n=4) and 2 (n=12) contained surveillance isolates from community sources predominantly. All but one of the clinical infections isolates belonged to clusters 3 (n=66) and 4 (n=10), respectively. On the basis of our calculations of the identification factors of VSEF and representative isolates of different VREF genogroups, we found that isolates of cluster 1 (n=4) resembled those of genogroup A, previously allocated to nonhospitalized patients and pig-derived VREF (Table 2). Similarly, isolates of cluster 2 (n=12) fitted best in genogroup B. Isolates from cluster 3 (n=66) showed almost equal resemblance to isolates from genogroups B and C, and isolates from cluster 4 (n=10) were also most identical to isolates from genogroup C (Table 2). The relationship between AFLP clusters of VSEF and sources was congruent with the previously described clustering of VREF. Almost all VSEF isolates (99%) derived from clinical infections clustered in clusters 3 and 4, clearly distinct from most VSEF isolates from community surveys (93%), which were found in clusters 1 and 2. A similar distribution was found previously among VREF with most (60%) isolates from clinical infections in genogroup C and most (89%) isolates from community surveys in genogroup A (11). The presence of the variant esp gene in VREF and VSEF was strongly associated with a specific epidemiologic source because the presence of esp is higher in clinical infections and epidemic-associated isolates than in surveillance isolates (Figure 2). VREF isolates associated with nosocomial outbreaks were esp-positive, except for one. Prevalences of the variant esp gene in clinical infectious isolates were 57% and 40% for VSEF and VREF, respectively (p=ns). Prevalence of the variant esp gene in clinical and community survey isolates was low among VREF (6% and 3%, respectively) and completely absent among the 19 VSEF isolates tested. Associations similar to the variant esp gene were found between ampicillin resistance and epidemiologic source for enterococcal isolates (Figure 2). All isolated associated with nosocomial VREF-outbreaks but one were resistant to ampicillin, as were 81% and 65% of infectious isolates of VSEF and VREF, respectively. Thirty-one percent of 36 nosocomial surveillance isolates of VREF were ampicillin resistant, as compared to none of five VSEF isolates obtained by clinical surveillance (p=ns). Finally, all but one isolate of VREF (n=36) and all VSEF isolates (n=14) obtained by surveillance of healthy persons were susceptible to ampicillin. When we combined these data, we found strong associations between the presence of the variant esp-gene and ampicillin resistance, both in VREF and VSEF: 98% of esp-positive VSEF and 92% of esp-positive VREF were resistant to ampicillin, as compared to 37% esp-negative VSEF and 20% esp-negative VREF isolates (p<0.0001). The purK housekeeping gene was sequenced in 103 isolates: 64 VREF and 39 VSEF. The previously described type 1 allele was found in 39 isolates: 23 VREF and 16 VSEF. This specific allele type was associated with the presence of the variant esp-gene and ampicillin resistance but not with vancomycin resistance. The variant esp-gene was found in 25 (64%) of 39 isolates containing the purK type 1 allele and in only 1 (2%) of 64 isolates carrying other purK alleles (p<0.0001). Similarly, ampicillin resistance was detected in 36 (92%) of 39 isolates with purK type 1 allele and in only 5 (8%) of 64 isolates with other allele types (p<0.0001). In contrast, the vanA transposon was present in 23 (59%) of 39 isolates with purK type 1 allele and in 41 (64%) of 64 isolates with other allele types (p=ns). DiscussionOur study demonstrates the genetic relatedness of clusters of isolates of vancomycin-resistant and -susceptible E. faecium strains from different epidemiologic sources and provides evidence for selection of an E. faecium subtype associated with hospital outbreaks. This subtype is characterized by the presence of both ampicillin resistance and the variant esp gene. Furthermore, our findings suggest random horizontal spread of the vanA transposon to multiple genogroups of E. faecium. We hypothesize that the rise in infections caused by VREF resulted from nosocomial selection of a specific ampicillin-resistant E. faecium genotype harboring the variant esp gene and subsequent horizontal transfer of the vanA transposon. Our study confirms that the previously demonstrated dichotomy between VREF isolated from healthy persons and patients (11) also exists for vancomcyin-susceptible E. faecium isolates. In VREF isolates, we could identify four genogroups, which were associated with particular hosts and environments and in which most isolates from healthy persons clustered distinctly from patient isolates. We showed that vancomycin-susceptible isolates clustered into three of these groups and that VSEF isolates from healthy persons also clustered distinctly from patient isolates. The genetic relationship between isolates and the genetic distinction between the four genogroups were based on AFLP analysis. We recently confirmed these findings with multilocus sequence typing (MLST) (13). Other researchers have also demonstrated host specificity of E. faecium. Quednau et al. suggested host specificity of isolates from chicken, pork, and humans by comparing restriction endonuclease profiles (26). In contrast, a recent study by Vancanneyt et al. also used AFLP; they did not confirm host-specific clustering of E. faecium (27). They described two main genomic groups in a population of 78 E. faecium strains isolated from seven European countries, and both groups comprised strains from (healthy) humans, animals, and food. All human clinical strains clustered in the largest genogroup, as did all strains (n=16) containing the vanA gene. Our findings that the vanA transposon was present in isolates of all genogroups proves that acquisition of this transposon was not influenced by and did not affect the preexisting relationship between the bacterium and host, which is a result of a long-term coevolution and mutual adaptation. The presence of four genogroups in E. faecium seems to parallel the phylogenetic structure in E. coli, in which four ancestral groups (A, B1, B2, and D) have been described with some level of host specificity (28). However, in contrast to our finding of a single genetic lineage in E. faecium related to clinical symptoms and carrying the esp virulence gene, clinical isolates of E. coli are more widely distributed among the different ancestral groups (29). Furthermore, MLST of pathogenic E. coli strains showed that different ancestral lineages have acquired the same virulence factors (30), indicating that pathogenic potential in E. coli is not confined to a single ancestral lineage, which is suggested by our findings for E. faecium. Human and animal pathogenic E. coli strains share closely related genotypes and carry similar virulence factor profiles, suggesting that certain E. coli strains are pathogenic for both animals and humans (31). Whether this holds true for pathogenic E. faecium strains is unknown. We recently found that the presence of the variant esp-gene is associated with nosocomial outbreaks of VREF in three continents, although this gene was not found in VREF strains isolated from healthy persons or animals (12). The outbreak strains were also characterized by a specific allele type of the purK gene, one of the housekeeping genes sequenced in the MLST method (13). Recently, other investigators reported the presence of the variant esp gene in clinical isolates of VSEF, demonstrating that this gene is not linked specifically to the vanA transposon (15–18). The findings of our study confirm the strong association between the presence of the esp gene and the relation with hospital outbreaks and clinical infections among patients with VREF as well as VSEF. Although the esp gene is virtually absent among community isolates, the presence of esp among VSEF and VREF from clinical infectious sites apparently unrelated to hospital outbreaks implies that this gene is not exclusively related to epidemic strains. Excluding the outbreak potential of the esp-positive VSEF strains in this and other studies is difficult. Only few outbreaks with VSEF have been documented (32–34). We have investigated and could not demonstrate the presence of the variant esp gene in isolates from one hospital outbreak of ampicillin-resistant E. faecium in Norway (data not shown). Little is known about the function of the variant esp-gene, although epidemiologic findings support its role as a virulence factor. In E. faecalis, the homologue of this gene encodes for the enterococcal surface protein, and the presence of this gene has been associated with enhanced adherence capacities to uroepithelial surfaces, but not with increased virulence, in a mice model (35). Moreover, in E. faecalis, the esp-gene was highly associated with biofilm-formation capacity (36). Increased adherence capacities and biofilm formation of esp-positive E. faecium strains might explain its association with hospital outbreaks. Like the variant esp gene, ampicillin resistance was found more frequently in isolates associated with infections and nosocomial outbreaks, both in VSEF and VREF. This source-relationship is probably caused by selective pressure of β-lactam antibiotics that are used extensively in hospitals. Emergence of ampicillin resistance in E. faecium was already demonstrated in the early 1980s and seemed to precede the emergence of vancomycin resistance by 10 years (2). A correlation between high prevalences of the esp gene and antibiotic resistance among E. faecium isolates from hospitalized patients was also reported recently by Coque et al. (17). Considering the sources of isolates, presence of ampicillin and vancomycin resistance, the presence of the variant esp gene, and the type 1 allele of the purK gene, we propose an evolutionary scheme for the specific genogroup of E. faecium associated with nosocomial outbreaks (Figure 3). However, our findings might have been biased by the composition of our collection of isolates and the fact that the purK was sequenced in a subset of all isolates. The esp gene and ampicillin resistance can obviously co-occur in a distinct genetic lineage of E. faecium characterized by the type 1 allele of the purK gene. We propose that E. faecium strains containing the type 1 allele of the purK gene have acquired the esp virulence gene and that this E. faecium genotype (purK-1, esp-positive) is prominently present among clinical relevant strains and virtually absent among survey isolates. Yet another substantial part of the clinical relevant strains with genotype purK-1 do not carry the esp gene, which emphasizes that other virulence genes of E. faecium apart from esp are involved in the development of infections. Ampicillin resistance was predominantly found among the purK-1 genotype and is almost absent among other E. faecium genotypes. This occurrence of resistance is, presumably, the result of selective antibiotic pressure. Chromosomal linkage of the purK-1 allele, the variant esp gene, and ampicillin resistance could have promoted this selection. Finally, glycopeptide usage in and outside hospitals, both in humans and animals, resulted in the selection of vancomycin-resistant strains in both the purK-1 genotype and the other genotypes. The presence of similar proportions of vancomycin resistance in all genotypes probably reflects horizontal transfer of the vancomycin-resistance transposon. This hypothesis implies the development of a hospital-adapted genogroup of E. faecium, characterized by the type-1 allele of purK, the variant esp-gene, and ampicillin resistance, which has spread unnoticed, thereby creating a pool of strains with epidemic potential. Only after becoming vancomycin-resistant has this genogroup become recognized as clinically relevant. Acknowledgments
References
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||
|
||||||||||||
|
|
|
EID Home | Top of Page | Ahead-of-Print | Past Issues | Suggested Citation | EID Search | Contact Us | Accessibility | Privacy Policy Notice | CDC Home | CDC Search | Health Topics A-Z |
||
|
This page posted August
1, 2003 |
||
|
Emerging
Infectious Diseases Journal |
||