 |
 |
 |
|
 |
FDA Home Page | Search FDA Site | FDA A-Z Index | Contact FDA
Department of Health and Human Services
U.S. Food and Drug Administration
May 2007
Message from Dr. Janet Woodcock
Council Mission Statement And Organization
Where To Find More Information
Appendix
2: Integrated Systems Approach to CMC Review and CGMP Inspection
4: Investigational Clinical Supplies
5: Manufacturing Science and Continuous Improvement
6: Part II - Electronic Records Requirements
7: Pharmaceutical Quality Standards
8: Process Analytical Technology
10: Quality Systems Continuous Improvement
11: Quality Systems Implementation - Pharmaceutical Inspectorate
12: Quality Systems Implementation - Recalls
13: Quality Systems Implementation - Warning Letters
14: Risk Management
16: Pharmaceutical Research Manufacturing Project
17: Workshops
18: Pharmaceutical Inspectorate (PI) Training Program
19: Quality by Design Chart Graphic
20: Acronyms Used
![]()
Working on the Pharmaceutical Quality for the 21st Century has been a rewarding experience for all of us at the Food and Drug Administration (FDA). Not only have we made a great deal of progress in modernizing our regulatory processes, we have learned much about improving pharmaceutical product quality that has helped us to develop flexible regulatory approaches to support continuous improvement in those processes. I am extremely proud of the progress we have made since we began this Initiative in 2002. Although we reported out much of that progress in 2004, I did not want anyone to lose sight of the need to continue to improve in the area of product quality and of FDA's continued efforts. The intent of this report is to bring everyone up-to-date on our activities and to ensure that the momentum of this extremely important Initiative is maintained.
Many improvements were made in the CGMP process at the beginning of the Initiative. Companies will agree that the focus of inspections has improved and that questions and concerns are addressed earlyier in the process. We continue to improve the inspectional process as well as enhance consistency across programs both in the field and in the affected Centers. Although we continue to focus on various aspects of the CGMP program for improvement, the last two years, our efforts have been directed to building and maintaining a strong science based review process, which can provide more synergism across all programs.
As you know, FDA cannot meet the goals of the Initiative alone. The success of the Initiative has been predicated on active participation and input from experts in industry, academia, government, and consumer groups. I want to thank everyone who has worked closely with us over the last few years by participating in workshops and conferences,, by helping to better define our direction for the Initiative, and by educating us at FDA on manufacturing science and the other important aspects of improving the regulatory programs.
The journey has just begun. There is still much to learn and innovations to incorporate into all our processes. We, in the agency, will continue to emphasize the importance of the Initiative and look forward to many more improvements in our regulatory processes for ensuring product quality.
To facilitate FDA's modernization of the regulation of pharmaceutical manufacturing and product quality, the FDA Management Council establishes a Council on Pharmaceutical Quality (herein referred to as Council). The Council serves as the guiding body on activities and policy development related to the modernization of the regulation of cross-center and Office of Regulatory Affairs (ORA) pharmaceutical manufacturing and product quality. The Council also serves as a resource to the FDA Management Council and to the agency in general on matters relevant to this subject.

FDA continues to make progress under the Pharmaceutical Quality for the 21st Century – A Risked Based Approach formally known as Pharmaceutical CGMP Initiative for the 21st Century – a Risk Based Approach. This Initiative, introduced by FDA in 2002, was intended to modernize FDA's regulation of pharmaceutical quality for veterinary and human drugs and select human biological products such as vaccines. The name has been changed to capture the larger issue of product quality, with CGMPs being an important tool towards improving overall product quality. The following organizational components of FDA have been actively involved in working toward this modernization over the last four years: ORA, Center for Biologics Evaluation and Research (CBER), Center for Drug Evaluation (CDER) and the Center for Veterinary Medicine (CVM), along with various offices within the Office of the Commissioner. All of these organizations continue to promote the goals and objectives of the Initiative and are dedicated to continuing to improve the regulation of pharmaceuticals in the 21st Century.
This report is intended to provide an update on the Agency's progress since the last report issued in September 2004 and to impress upon the pharmaceutical industry and our stakeholders the continued importance of this Initiative to the Agency.
The Initiative was originially issued with the following goals:
This report and the report issued in September 2004 specify the accomplishments that were made in achieving the goals of the Initiative. This report will set forth the current activities of the Agency as well as inform the public of the future direction of the Agency in implementing the goals of the Initiative.
This report highlights those general activities and projects of the Initiative that have been worked on under the auspices of the Council in the last two years. Over the last two years, the Council has identified additional objectives for continuing to improve drug quality regulation based on new challenges and the need for continuous improvement of the regulatory programs. A number of additional multidiscipline work groups have been established under the Council to focus on these activities. The current work groups are listed in the Organization Section of this report. Individual reports from all of the work groups are also attached which provide specific information on accomplishments and future goals of the each work group.
Communication is an important aspect of implementing the Initiative and falls under the responsibilities of the Council. Besides organizing and/or participating in workshops both internally and externally to communicate changes in regulatory programs and to solicit information from industry and other stakeholders, several new guidances or draft guidances have been issued in the last two years to support the goals of the Initiative.
The Agency held the following workshops to discuss the modernization of regulatory programs since the last report on the Initiative:
See Appendix 17 for more information on several of the workshops.
The following final and draft guidances issued since the last report:
Included in the responsibilities of the Council is overseeing the ongoing implementation of internal quality management systems. The Council has provided guidance and direction for all activities involving drug quality and has worked closely with external groups to understand stakeholder needs and related initiatives. Under the direction and oversight of the Council, several internal quality management systems have been or are being established. This includes developing internal quality systems for processing recalls and warning letters to facilitate internal procedures and for oversight and management of the Pharmaceutical Inspectorate (PI) in order to create better consistency and regulatory effectiveness of inspections. Work has started on developing a performance-based quality management system for the CMC review process and the laboratory-based product quality programs across CDER and CBER. CVM has implemented a quality management system in the past two years for its CMC review.
In order to meet the goal of the Initiative to facilitate industry's adoption of modern quality systems, FDA published a guidance on quality systems approach to pharmaceutical CGMPs. This guidance not only provides information to help in implementing quality systems and risk management approaches but also provides the framework for integrating these approaches into existing programs with the goal of encouraging industry to adopt modern and innovative manufacturing technologies.
Collaboration with international health and regulatory organizations has been a vital part of the modernization efforts. The focus has been towards promoting international cooperation directed at assuring drug product quality and consistency of CGMPs. Ongoing activities include:
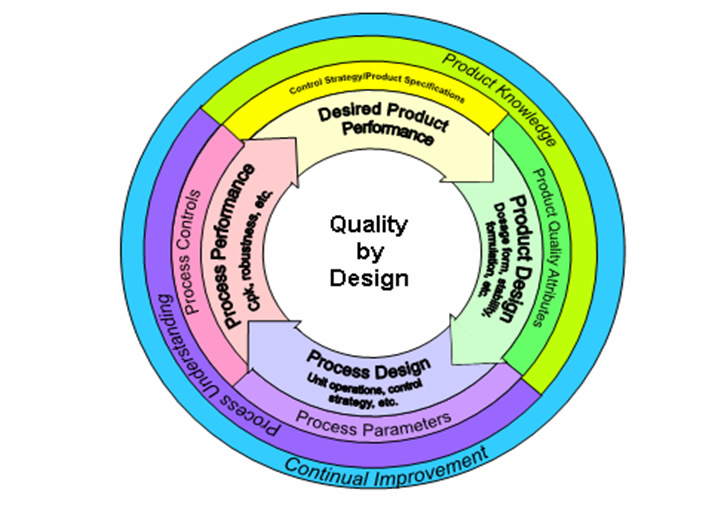
Significant progress has been made in the implementation of the concepts of "Quality by Design" (QbD) into Agency processes. The focus of this concept is that quality should be built into a product with a thorough understanding of the product and process by which it is developed and manufactured along with a knowledge of the risks involved in manufacturing the product and how best to mitigate those risks. The chart in Appendix 19 demonstrates the essential aspects of QbD. Specifically, the following activities are being highlighted as being part of the overall implementation of QbD:
Implementation of Process Analytical Technologies (PAT) as a tool for designing
and analyzing pharmaceutical development and manufacturing is an important
concept for meeting the goal of the Initiative of encouraging new manufacturing
technologies. The Agency has been engaged in review of a number
of applications for both new drugs and generic drug products in which companies
are applying technologies, which are currently available to provide information
on physical, chemical, (micro) biological characteristics of materials to
improve process understanding and to measure, control, and/or predict quality
and performance of products.
There have also been a number of training programs, both internally and externally,
to highlight the applicability of PAT and the regulatory pathways. In
addition, an executive committee was established in ASTM (E55) to develop standards
for implementation of PAT in pharmaceutical manufacturing.
The PI was established in the ORA to enhance the Agency's overall inspection program. The following progress has been made in ensuring PI success:
See Appendix 18 for additional information on the PI Training Program
Scientific collaboration continues to be an important part of accomplishing the goals of the objectives. The Agency continues to be very active in its collaboration efforts with external constituents from both academia and industry. The collaborative efforts are also essential in meeting the critical path needs. There are a great many challenges, which exist in drug development and manufacturing that are being tackled while accomplishing the goals of the 21st Century Initiative. Many of the work groups under the Council are actively involved in developing or maintaining collaborations. The Council has also met with a number of outside organizations to understand what is happening in various areas that coincide with the efforts of the Council.
Examples of some of these collaborative efforts are:
Many of these collaborations also are significant elements of the Critical Path initiative that was established to focus on the modernization of techniques and methods used to evaluate safety, efficacy and quality of medical products. In enhancing the regulatory processes for ensuring quality pharmaceutical products, a number of research projects are being done in collaboration with industry and academia to improve manufacturing science.
The Council supports Office of Management and Budget (OMB) Circular A-119, which establishes policy on use and development of voluntary consensus standards. The tenets of the 21st Century Initiative also emphasize the need to use consensus standards to support the regulation of pharmaceuticals. The Initiative promotes using standards for (1) understanding new technologies, (2) ensuring consistency across industry and Agency, (3) optimizing efficiency in regulating, and (4) promoting international harmonization. The Council has established a work group to determine how best to support and utilize standards across the pharmaceutical quality programs.
As FDA is a strong proponent of the use of standards, it encourages the pharmaceutical industry and other stakeholders to participate in standards-setting organizations where development of standards will be beneficial to the manufacture of high quality pharmaceuticals. FDA's primary role is to determine which standards, if accepted during application review, would support a more efficient review process and enhanced product quality assurance. Through guidances and other external communications, FDA will acknowledge the applicability of specific standards. Industry is encouraged to use and cite appropriate standards in their submissions. FDA will access the suitability of cited standards within the context of the manufacturer's application.
FDA has also participated with ASTM in developing standards for implementing PAT. These standards will help the Agency have a better understanding of the technology and provide the industry with guidance on implementing PAT into their manufacturing processes.
FDA continues to facilitate the modernization of regulatory processes related to pharmaceutical products in the 21st Century. This report is being issued to emphasize the Agency's commitment to the goals and objectives in pursuit of more consistent and effective regulatory processes. The attached reports provide the public with a general idea of many of the Agency's activities in pursuing these goals and objectives.
Separate guidance page:
Questions
Direct any questions on this report to Ted Sherwood who serves as the Project Manager for the Council of Pharmaceutical Quality.
Attention: Ted Sherwood
FDA, CDER, OPS, IO
10903 New Hampshire Ave.
Bldg 21, Room 3528
Silver Spring MD 20903-0002
Tel: 301-796-1605
Fax: 301-796-9733
email: edward.sherwood@fda.hhs.gov
![]()
Chairs: Fred Blumenschein and Mary Malarkey
Project
Manager: Vikki Kinsey
Members: Pat Alcock, ORA, Diane Alexander, CBER, Neal Bataller, CVM, , Fred Blumenschein, CDER, Walt Brown, ORA, James Cohen, OCP, Mary Davis-Lopez, CBER, Kevin Fain, OCC, , Jo Gulley, CVM, Brian Hasselbalch, CDER, Vikki Kinsey, CDER, Joan Loreng, ORA, , Mary Malarkey, CBER, Sharon Mayl, OPPL, Grace McNally, CDER, James Nitao, CVM, Charles O'Brien, CVM, LuAnn Pallas, ORA, Michael Rogers, ORA, Jessica Tave, OCBQ
Background
The CGMP Regulations Work Group was formed to implement modifications to 21 CFR Part 210/211. As described in the September 2004 Final Report "FDA will take an incremental approach to modifying parts 210/211, while pursuing international harmonization through ICH and PIC/S."
Goal(s) and Tasks
INTEGRATED SYSTEMS APPROACH TO CMC REVIEW AND CGMP INSPECTION (INTERACTION) WORK GROUP
Chair: Chris Watts
Facilitator: Sarah Mullikin (SVM Consulting)
Members: Julie Conwell, CVM, Gwyn Dickinson, ORA, Susan Pittinger, CDER, Sarah Pope, CDER, Edwin Rivera-Martinez, CDER, Nancy Rolli, ORA/NJ-DO, Robin Stone, CVM, Chris Watts, CDER
Background
A primary objective of the quality initiative is to "enhance the consistency and predictability of FDA's approach to assuring product quality and safety among the FDA's centers and field components." In order to realize this objective, among other activities, the Council established this work group with the charge to improve the interaction between CMC review and CGMP inspection, thereby facilitating a consistent regulatory approach to product and process quality across various Agency components.
Goal(s) and Tasks
![]()
INTERNATIONAL ACTIVITIES WORK GROUP
Chair: Lois Ann Beaver
Project Manager: Tammie
Bell
Members: Lois Beaver, OC, Dennis Bensley, CVM, Joan
Blair, CBER, Monica Caphart, CDER, Katherine Cooper, OC, Doug
Ellsworth, ORA, Joe Famulare, CDER, Sema Hashemi, OC, Brian Hasselbalch, CDER,
Diana Kolaitis, ORA, Michelle Limoli, OC, Scott MacIntire, ORA, Justina Molzon,
CDER, Anita Richardson, CBER, Michael Rogers, ORA, Merton Smith, CVM,
Steve Solomon, ORA
Background
Developing harmonized international scientific standards is a continuing activity, with the FDA actively collaborating with other regulatory authorities, in multilateral, international forums, such as the International Conference on Harmonization of the Technical Requirements for Registration of Pharmaceuticals (ICH) and the International Cooperation on Harmonization of Technical Requirements for Registration of Veterinary Medicinal Products (VICH). This Work Group leads efforts to promote international regulatory cooperation directed towards assuring drug product quality and GMPs
Goal(s) and Tasks
INVESTIGATIONAL CLINICAL SUPPLIES WORK GROUP
Chairs: Joe Famulare and Chris Joneckis
Project
Manager: Vacant
Members: Monica Caphart, CDER, John Dietrick, CDER, Joe Famulare, CDER, Chris Joneckis, CBER, Laurie Norwood, CBER, Walt Brown, ORA, Guraig Poochikian, CDER, Chiang Syin, CBER, Dan Takefman, CDER, Brenda Uratani, CDER, Keith Webber, CDER, Paula McKever, OC, Peter Beckerman, OC, Rachel Berman, OC,
Background
In response to comments from regulated industry and academic/ clinical developers, FDA recognized that application of the CGMP regulations, as described in 21 Code of Federal Regulations CFR 211, is not always relevant for the manufacture of clinical investigational drug products. Furthermore, FDA's current approach of employing enforcement discretion in applying CGMPs at 21 CFR 211 was not acceptable to manufacturers of clinical investigational drug products because of the concern of the legal liability to comply with all of the GMP regulations specified in 21 CFR 211. In response to this and internally-driven needs for clarification, the Agency recognized the need to develop specific GMPs for investigational products. FDA elected to address the progressive nature of CGMPs in drug development for a wide variety of manufacturing situations and product types by providing appropriate guidance for compliance with CGMPs starting with Phase 1 studies.
Goal(s) and Tasks
![]()
MANUFACTURING SCIENCE AND CONTINUOUS IMPROVEMENT WORK GROUP
Chair: Moheb Nasr
Project Manager: Christine Moore
Members: Diana Amador, ORA, William Bargo, CVM, Andrew Chang, CBER, Jon Clark, CDER, Robert Coleman, ORA, John Dietrick, CDER, Douglas Ellsworth, ORA, Raafat Fahmy, CVM, Joseph Famulare, CDER, Frank Holcombe, CDER, Mai Huynh, CVM, Chris Joneckis, CBER, Diana Kolaitis, ORA, Steven Kozlowski, CDER, See Yan Lam, CDER, Grace McNally, CDER, Elaine Morefield, CDER, Elise Murphy, ORA, Guirag Poochikian, CDER, Vilayat Sayeed, CDER, Chris Watts, CDER, Keith Webber, CDER, Mansoor Khan, CDER
Background
The Manufacturing Science and Continuous Improvement Work Group provides a forum for discussion of FDA positions and coordination of activities regarding pharmaceutical manufacturing. The work group proposes activities and recommendations to the Council of Pharmaceutical Quality.
Goals and Accomplishments
PART 11 - ELECTRONIC RECORDS REQUIREMENTS WORK GROUP
Chair: Joe Famulare
Project Manager: Diane
Hanner
Members: Beers Block, Patricia; Carvajal, Ricardo; Doleski, Joseph (David); Druckman, Michael; Elder, David K.; Epstein, Laura; Famulare, Joseph; Fitzgerald, Brian; Gallant, David; Gatling, Bob; Goldstein, Jennifer; Hanner, Diane; Henrikson, Erik N; Lienesch, John; Loreng, Joan; Mednick, David; Murray, John F.; Sims, Marguerita; Smith, George; Thomas, Audrey A; Thomas, Jennifer; Toelle, Vernon D; Wyn, Sion; Zabriski, Margaret A; Ziyad, JoAnn
Background
As an outgrowth of its quality initiative for human and animal drugs
and biologics, FDA continues to re-examining Part 11 as it applies to
all FDA regulated products. Part 11 regulations provide criteria for acceptance
by FDA, under certain circumstances, of electronic records, electronic signatures,
and handwritten signatures executed to electronic records as equivalent to
paper records and handwritten signatures executed on paper. These regulations,
which apply to all FDA program areas, were intended to permit the widest possible
use of electronic technology, compatible with FDA's responsibility to protect
the public health.
Goal(s) and Tasks
PHARMACEUTICAL QUALITY STANDARDS WORK GROUP
Chair: Helen Winkle
Project Manager: Chris Watts
Members: Gary Buehler, CDER, Jon Clark, CDER, Joseph Famulare, CDER, Rick Friedman, CDER, Frank Holcombe, Jr, CDER, Chris Joneckis, CBER, David Kelly, OC, Steven Kozlowski, CDER, Donald Marlowe, OC, William Marnane, CVM, Steve Niedelman, ORA, Charles O'Brien, CVM, Guiragos Poochikian, CDER, Herman Schoenemann, CVM, Chris Watts, CDER, Keith Webber, CDER, Helen Winkle, CDER
Background
This work group was formed to develop and implement a process for evaluating and facilitating the use of standards related to pharmaceuticals and pharmaceutical manufacturing.
Goal(s) and Tasks
The goals of this work group are:
PROCESS ANALYTICAL TECHNOLOGY WORK GROUP
Chair: Helen Winkle
Project Manager: Chris Watts
Members: Ali Afnan, CDER, Patricia Alcock, ORA, Dennis Bensley, CVM, Jon Clark, CDER, Doug Ellsworth, ORA, Joe Famulare, CDER, Frank Holcombe, CDER, Chris Joneckis, CBER, Steve Kozlowski, CDER, Moheb Nasr, CDER, Chris Watts, CDER, Keith Webber, CDER, Helen Winkle, CDER
Background
The goal of PAT is to understand and control the manufacturing process, which is consistent with our current drug quality system: quality cannot be tested into products; it should be built-in or should be by design.
A desired goal of the PAT framework is to design and develop processes that can consistently ensure a predefined quality at the end of the manufacturing process. Such procedures would be consistent with the basic tenet of quality by design and could reduce risks to quality and regulatory concerns while improving efficiency. Gains in quality, safety and/or efficiency will vary depending on the product and are likely to come from: 1) reducing production cycle times by using on-, in-, and/or at-line measurements and controls; 2) preventing rejects, scrap, and re-processing; 3) real- time release; 4) increasing automation to improve operator safety and reduce human error; 5) facilitating continuous processing to improve efficiency and manage variability using small-scale equipment (to eliminate certain scale-up issues) and dedicated manufacturing facilities, improving energy and material use, and increasing capacity.
Goal(s) and Tasks
![]()
PROCESS VALIDATION WORK GROUP
Chairs: Brian Hasselbalch and Grace McNally
Project Manager: As above
Members: Brian Hasselbalch, CDER, Ali Afnan, CDER, John Dietrick, CDER, Grace McNally, CDER, Evans, CDER, Chris Watts, CDER, Elaine Morefield, CDER, Mary Malarkey, CBER, Chris Joneckis, CBER, John Finkbohner, CBER, William Marnane, CVM, Dennis Bensley, CVM, Jan Welch, CDRH, Daniel Grabicki, ORA, Thomas Arista, ORA
Background
This group was formed to revise the 1987 guidance, General Principles of Process Validation.
Goal(s) and Tasks
QUALITY SYSTEMS CONTINUOUS IMPROVEMENT WORK GROUP
Chairs: Robert Sausville and
Joseph Famulare
Project Manager: Patrick Guinn
Members: Diane Alexander, CBER, Diana Amador, ORA, Dennis Bensley, CVM, Monica Caphart, CDER, Chris Joneckis, CBER, June Liang, CVM, Patricia Maroney-Benassi, ORA, Sherry Purvis-Wynn, ORA, Michael Smedley, CDER, Joe Famulare, CDER, Robert Sausville, CBER, Patrick Guinn, CDER
Background
The Quality System Guidance Development work group (QS work group) is responsible for comparing the current CGMP regulations, which call for specific quality management elements, to other existing quality management systems. The QS work group mapped the relationship between CGMP regulations (parts 210 and 211 and the 1978 Preamble to the CGMP regulations) and various quality system models such as the Drug Manufacturing Inspections Program (i.e. systems-based inspectional program), the Environmental Protection Agency's Guidance for Developing Quality Systems for Environmental Programs, International Standards Organization (ISO) Quality Standards, other quality publications, and experience from regulatory cases.
Goal(s) and Tasks
OFFICE OF REGULATORY AFFAIRS PHARMACEUTICAL INSPECTORATE QUALITY MANAGEMENT SYSTEMS TASK FORCE
Chair: Susan Setterberg
Project Manager: Eileen
Cole
Members: Susan Setterberg, ORA, Helen Winkle, CDER, Kristen Anderson, CVM, Chuck Edwards, ORA, Douglas Ellsworth, ORA, Kristen Evans, CDER, David Horowitz, ORA, Yvette Johnson, ORA, PHI-DO, June Liang, CVM, Patricia Maroney-Benassi, ORA, Nancy Rolli, ORA, NJ-DO, John Thorsky, ORA, SW/KAN-DO, Chris Watts, CDER, Faye Wei, CVM
Background
The ORA PI QMS Task Force was established to develop a quality management system
for the Pharmaceutical Inspectorate (PI). The prime focus is to identify
issues relating to developing a quality management system, ensure interaction
between reviewers and inspectors, make more information available on the
PI, and discuss how the PI will be used in the future.
Goal(s) and Tasks
Create an environment that ensures quality, consistency, and regulatory effectiveness of inspections so that the reviewers and investigators have the same common goals
![]()
QUALITY SYSTEMS IMPLEMENTATION WORK GROUP - RECALLS
Chairs: David Elder and Carl Draper
Project
Manager: Patricia Maroney-Benassi
Members (Recall QS Project Team): Anna Marie Kempic, Office of Chief Counsel, Dorothy Miller, Office of Crisis Management, Catherine McDermott, OER/OPA, Media Relations Staff, Isadora Stehlin, OER/OPA, Website Mgt. Staff, Len Valenti, OIASI/International Programs, Dianne Murphy, OIASI/ Pediatric Therapeutics, Mark Kramer, OIASI/Combination Products, Laura Hieronymus, CBER, Lavonia Huff, CDER, Frances Benedict, CDRH, Liliane Brown, CDRH, Cecilia Wolyniak, CFSAN, Barbara Rodgers, CVM, Glenn Peterson, CVM, Bradford Stone, ORA, Mel Szymanski, ORA/Enforcement, Sherrie Krolczyk, ORA/Southwest Region
Background
One goal of the FDA quality Initiative is to integrate quality systems and risk management approaches into existing agency programs. The Initiative's members (CBER, CDER, CVM, and ORA) agreed with the Center for Devices and Radiological Health (CDRH) and Center for Food Safety and Applied Nutrition (CFSAN) to develop agency-wide quality system implementation of two specific projects focused on recalls and warning letters.
Goal(s) and Tasks
QUALITY SYSTEMS IMPLEMENTATION WORK GROUP - WARNING LETTERS
Chairs: Carl Draper and David Elder
Project Officer: Valerie Vallely
Project Manager: Vacant
Members: Joy Dawson, Office of Chief Counsel, M. Dianne Murphy, Office of Pediatric Therapeutics, Mark Kramer, Office of Combination Products, Rosie Whitcraft, Office of the Chief Information Officer, Fred Sadler, Office of Management Programs, Anita Richardson, CBER, Frances Benedict, CDRH, Liliane Brown, CDRH, Deborah Autor, CDER, Leslie Ball, CDER, Lesley Frank, CDER, Judith Gushee, CFSAN, Mark Hackman, CVM, James Kewley, ORA/Field
Background
One of the Agency's goals is the integration of quality systems and risk management approaches into existing Agency programs and processes. The Warning Letter process was identified as a quality system candidate because of its significance in achieving industry compliance and in emphasizing the importance of timeliness, consistency, and legal sufficiency in the final work product.
Goal(s) and Tasks
![]()
RISK MANAGEMENT WORK GROUP
Chair: David Horowitz, Deborah Autor
Project Manager: Vacant
Members: John Gardner, CDER, Brenda Wang, CDER, Vincent Zenger, CDER, Suzanne Barone, CDER, Karen Kirchberg, CDER, Nicholas Buhay, CDER, George Smith, CDER, Jon Clark, CDER, Vilayat Sayeed, CDER, Charles Gray, CVM, Meyer Slobotsky, ORA, NJ-DO, Maridalia Torez-Irizarry, ORA, Stephen Souza, ORA, NEW-DO
Background
The group was initially formed to explore opportunities for applying risk-based approaches to prioritize and focus the various activities performed by FDA concerning the oversight of GMP requirements. The group then concentrated its efforts on developing and implementing a quantitative model to prioritize inspections of drug manufacturing facilities. The highest priority sites selected by the model are counted toward the agency's annual performance goal for high-risk human drug inspections under the Government Performance Results Act. The work group developed and implemented the expert elicitation survey, which gathered data from agency experts to identify and weigh factors associated with (1) maintaining manufacturing process control and (2) vulnerability to cross-product or environmental contamination. The group also participated in drafting the white paper that described how the model would be piloted in FY ‘05 and in implementing that pilot. The work group next devoted its attention to improving and continuing implementation of the model. Significant advancements in FY ‘06 included data quality improvement efforts carried out by the field; the inclusion on data from Field Alert Reports associated with facilities; and on therapeutic risk categories associated with products.
Goal(s) and Tasks
DISPUTE RESOLUTION WORK GROUP
Chairs: David Horowitz and Helen Winkle
Project Manager: Ted
Sherwood
Members: Mike Rogers, ORA, Walt Brown, ORA, Doug Ellsworth, ORA, Mary Malarkey, CBER, Anita Richardson, CBER, Albi D'Sa, CDER, John Dietrick, CDER, Teddi Lopez, CDER, Jorge Christian, CVM, Mary Leadbetter, CVM
Background
The Agency acknowledged concern expressed by the pharmaceutical industry that disputes related to scientific and technical issues may arise during FDA inspections of pharmaceutical manufacturers to determine compliance with CGMP requirements or during the Agency's assessment of corrective actions undertaken because of such inspections. As these disputes may involve complex judgments and issues that are scientifically or technologically important, it is critical to have procedures in place that will encourage open, prompt discussion of disputes and activities leading to their resolution. The Agency also realized that industry uses the outcomes of inspections as tools to prepare for future inspections. Therefore, the Agency recognizes the importance of reporting the proper inspection outcomes. At the onset, it was clear that formal procedures for raising disputes to ORA and Center levels were lacking.
Goal(s) and Tasks
Please note: This Work Group completed its task and is no longer active.
PHARMACEUTICAL RESEARCH MANUFACTURING COLLABORATION
Conducted under an arrangement with Professor Jeff Macher of Georgetown University and Professor Jack Nickerson of Washington University, St. Louis; the Council received results generated from statistical analyses of data comprised of CDER, ORA and Industry data metrics.
These statistical analyses derived from two phases of the Professors' Pharmaceutical Research Manufacturing Project (PRMP). The first phase involved working with the FDA to collect and compile data from FDA internal databases. Based on this data, the project developed risk-based statistical models to assess the probability of a facility being chosen for inspection, to evaluate the effect of investigator training and experience on the probability of investigational outcomes as well as individual investigator effects on the probability of investigational outcomes, and to identify characteristics of facilities and firms that correlate with the likelihood of noncompliance. A preliminary report was delivered to the FDA on January 28, 2005.
The second phase of the PRMP focused on collecting and analyzing data from 42 pharmaceutical manufacturing facilities of 19 manufactures. Preliminary results were presented to FDA on February 17, 2006. While results from all 27 statistical analyses were presented in the report, five consistent findings were identified. First, the adoption of information technology by manufacturing facilities corresponds to higher manufacturing performance metrics. Second, the locus of decision rights, especially with respect to deviation management, affects performance metrics. Decision rights located closer to the shop floor generally improve manufacturing performance metrics.
Third, facilities engaged in contract manufacturing generally correspond to
worse performance metrics. Fourth, the use of process analytic technology tools
generally, although not in all instances, corresponds to worse performance
metrics. This correspondence, however, does not imply causation, which means
that these tools may be adopted because of poor performance metrics instead
of generating poor performance. Finally, the scale and scope of the manufacturing
facility have a complex interplay with manufacturing performance as both a
benefit and a detriment to performance depending on the metric of interest
and the type of production process.
Next steps involve the Professors working with CDRH to develop a risk-based
assessment tool. This tool will be implemented by the FDA to plan inspections
based on a facility's risk profile.
AAPS WORKSHOP ON PHARMACEUTICAL QUALITY ASSESSMENT - A SCIENCE AND RISK-BASED CMC APPROACH IN THE 21ST CENTURY
Co-sponsored with FDA and ISPE, October 5 - 7, 2005, North Bethesda,
Maryland
Summary
600 scientists and regulators from industry and government agencies around the world gathered to discuss concerns, make suggestions, and engage in challenging debate about the U.S. Food and Drug Administration's new science and risk-based pharmaceutical quality assessment system. The new assessment system is based on principles designed to facilitate regulatory flexibility, thereby allowing for a more efficient drug approval process without compromising product quality and efficacy. FDA's vision is to empower industry scientists to create development parameters, called "Design Space," using quality risk management principles and prior knowledge. Operating within a given product's "Design Space" assures a more efficient review process and eliminates the need for multiple submissions, which often slows down the drug approval process and delays the research from reaching its final and most important audience, the patient. The workshop planning committee consisted of the following individuals:
The basic format of the workshop was three morning plenary sessions that incorporated talks on Pharmaceutical Quality in the 21st Century, A New Pharmaceutical Quality Assessment System (PQAS), Understanding Key Terms, Quality by Design (QbD), Utilization of Comprehensive Quality Overall Summary (QOS) in CMC Submission and Review, Innovation and Continuous Improvement, and Challenges and Opportunities in Drug Development, Manufacturing, and Regulations. The plenary sessions were enhanced by breakout sessions and case studies on days 1 and 2 and an animated panel discussion on day 3.
University of Rhode Island (URI)
Conference
UNIVERSITY OF RHODE ISLAND (URI) CONFERENCE
ON CMC – 21ST CENTURY CHEMICAL MANUFACTURING AND CONTROLS STRATEGIES
Co-sponsored with FDA October 17 - 18, 2006, Reston Hyatt, Reston,
Virginia
Summary
The U.S. Food and Drug Administration (FDA) and the University of Rhode Island (URI), College of Pharmacy co-sponsored a workshop on the progress the FDA has made on the 21st Century Initiative since the final report. This event included discussions with the Director of each CMC review Office in CDER and the Director of the Office of Testing and Research. Details of specific efforts to better meet Critical Path Industrialization goals where provided.
FDA wanted to provide an open dialogue with the pharmaceutical industry in order to provide new critical information to them for leveraging advantages available in the Critical Path Industrialization and CGMPs for the 21st Century Initiatives. This venue provided an audience to focus on the implementation of these initiatives in areas specific to commercial manufacturing and the interaction of OPS and CGMP.
AAPS WORKSHOP ON PHARMACEUTICAL QUALITY ASSESSMENT - A MODERN RISK-BASED APPROACH
Co-sponsored with FDA and ISPE, February 28- March 2, 2007, Bethesda
North Marriott Hotel and Conference Center, North Bethesda, Maryland
Summary
This workshop was a follow up to the PQRI/FDA Workshop on A Drug Quality System for the 21st Century held in April 2003. It was being planned under the auspices of the Council on Pharmaceutical Quality at FDA and was co-sponsored with the American Association of Pharmaceutical Scientists and the International Society of Pharmaceutical Engineers. The 2-1/2 day program was intended to present progress on FDA's pharmaceutical quality initiatives. Furthermore, the workshop allowed regulated industry, other stakeholders, and the public to comment on progress made and to provide input to facilitate implementation of a common vision for pharmaceutical manufacturing in the 21st century. Among topics addressed: pharmaceutical development, chemistry, manufacturing and controls (CMC), manufacturing and quality operations, good manufacturing practices (GMP), quality systems and quality assurance. Over 500 scientists and regulators from industry and the FDA attended the workshop. The plenary sessions and the breakout sessions provided a number of recommendations which are now being reviewed by FDA and incorporated into current thinking.
PHARMACEUTICAL INSPECTORATE TRAINING PROGRAM
A significant milestone was reached on FDA's progress under the Pharmaceutical Inspectorate program during the past year (http://www.fda.gov/CDER/GMP/MOU.PDF). Fourteen of FDA's experienced investigators became the initial members of the Pharmaceutical Inspectorate upon completion of Level III certification. In addition, one CDER employee achieved Level III certification. With this achievement, FDA's systematic effort began to produce its expected results towards constantly upgrading the scientific, technical and regulatory knowledge base and skill sets of its field investigators.
FDA's Level III Drug Investigator Certification Board and its Course Advisory Group comprised of experienced members from CDER, CVM, and ORA have now established a well-structured program towards further enhancing the work efficiency of FDA's Drug Investigators. For the first cohort of PI members, a three-week didactic training was conducted at FDA's ORA University. The PI curriculum and the didactic courses consisted of topics such as:
After the didactic courses, each candidate participated in a four-week visit to CDER and CVM Offices responsible for the assessment of product quality and regulatory compliance, according to FDA's "Detail Rotation" program (http://www.fda.gov/cder/ops/PI-rotation.htm).
Each of our Level III Drug Investigators who are now members of the Pharmaceutical Inspectorate commented that it was a rewarding experience for them to go through the entire PI training program. They described the PI program to be very useful in our effort to increase efficiency of FDA's operations through extensive interaction between application review, site inspection programs, and monitoring of pharmaceutical manufacturers' compliance to applicable regulations. Because of the highly valued nature of the Pharmaceutical Inspectorate training they received, almost every investigator has recommended that the Agency should consider offering many elements of the Level III Drug Investigator training in the earlier phases of a Drug Investigator's training, for further improving FDA's capabilities to protect and promote public health. The Council will consider implementing their suggestions.
QUALITY BY DESIGN GRAPHIC
![]()
ACRONYMS USED
AAPS: American Association of Pharmaceutical Scientists
BLA: Biologics License Application
CBER: Center for Biologics Evaluation and Research
CDER: Center for Drug Evaluation and Research
CDRH: Center for Devices and Radiological Health
CFR: Code of Federal Regulations
CFSAN: Center for Food Safety and Applied Nutrition
CGMP: Current Good Manufacturing Practices
CMC: Chemistry, Manufacturing and Controls
CRADA: Cooperative Research and Development Agreement
CVM: Center for Veterinary Medicine
DFS: Division File System
DHRD: Division of Human Resource Development, Office of Resource Management, Office of Regulatory Affairs
EIR: Establishment Inspection Reports
FDA: Food and Drug Administration
FMD: Field Management Directives
ICH: International Conference on Harmonization
IND: Investigational New Drug
ISO: International Standards Organization
ISPE: International Society of Pharmaceutical Engineers
MOU: Memorandum of Understanding
NDA: New Drug Application
OMB: Office of Management and Budget
ONDQA: Office of New Drug Quality Assessments, Office of Pharmaceutical Science, Center for Drug Evaluation and Research
ORA: Office of Regulatory Affairs
PAT: Process Analytical Technologies
PI: Pharmaceutical Inspectorate
PIC/S: Pharmaceutical Inspection Cooperation Scheme
PQRI: Product Quality Research Institute
PRMP: Pharmaceutical Research Manufacturing Project
PQAS: Pharmaceutical Quality Assessment System
Q (ICH Q): Quality topic
Q4B: Regulatory Acceptance of Analytical Procedures and/or Acceptance Criteria
Q8: Pharmaceutical Development
Q9: Quality Risk Management
Q10: Pharmaceutical Quality Systems
QbD: Quality by Design
QMS: Quality Management System
QOS: Quality Overall Summary
QS: Quality Systems
URI: University of Rhode Island
USP: United States Pharmacopeia
VICH: International Cooperation on Harmonization of Technical Requirements for Registration of Veterinary Medicinal Products
![]()