About CJD
 |
 |
|
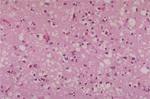
This tissue slide shows sponge-like lesions in
the brain tissue of a classic CJD patient. This lesion is typical of many
prion diseases. Larger
Picture
(Image courtesy Ermias Belay) |
|
Classic CJD is a human prion disease. It is a neurodegenerative disorder
with characteristic clinical and diagnostic features. This disease is rapidly
progressive and always fatal. Infection with this disease leads to death usually
within 1 year of onset of illness.
Important Note: Classic CJD is not
related to "mad cow" disease. Classic CJD also is distinct from "variant CJD",
another prion disease that is related to BSE.
 For information about these diseases, see: For information about these diseases, see:
Occurrence and Transmission
Classic CJD has been recognized since the early 1920s. The most common form of
classic CJD is believed to occur sporadically, caused by the spontaneous
transformation of normal prion proteins into abnormal prions. This sporadic disease
occurs worldwide, including the United States, at a rate of approximately one case
per 1 million population per year, although rates of up to two cases per million
are not unusual. The risk of CJD increases with age, and in persons aged over 50
years of age, the annual rate is approximately 3.4 cases per million. In recent
years, the United States has reported fewer than 300 cases of CJD a year.
Whereas the majority of cases of CJD (about 85%) occur as sporadic disease, a
smaller proportion of patients (5-15%) develop CJD because of inherited mutations
of the prion protein gene. These inherited forms include
Gerstmann-Straussler-Scheinker syndrome and fatal familial insomnia.
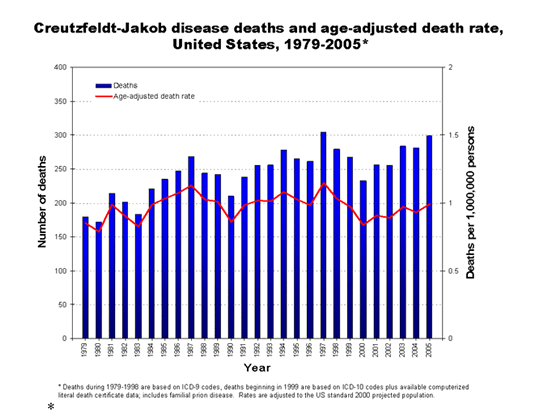 |
Select the graph for larger image
* Deaths during 1979-1998 are based on ICD-9 codes, deaths beginning in 1999 are based on ICD-10 codes plus available computerized literal death certificate data; includes familial prion disease. Rates are adjusted to the US standard 2000 projected population.
|
Clinical and Pathologic Characteristics of Classic CJD
Classic CJD characteristics, as compared to variant CJD, are presented in the
table below.
Clinical and Pathologic
Characteristics
Distinguishing Classic CJD from Variant CJD
| Characteristic |
Classic CJD |
Variant CJD |
| Median age at death |
68 years |
28 years |
| Median duration of illness |
4-5 months |
13-14 months |
| Clinical signs and symptoms |
Dementia; early neurologic signs |
Prominent psychiatric/behavioral symptoms; painful
dyesthesiasis; delayed neurologic signs |
| Periodic sharp waves on
electroencephalogram |
Often present |
Often absent |
| "Pulvinar sign" on MRI* |
Not reported |
Present in >75% of cases |
| Presence of "florid plaques" on
neuropathology |
Rare or absent |
Present in large numbers |
| Immunohitochemical analysis of brain
tissue |
Variable accumulation |
Marked accumulation of protease-resistance prion
protein |
| Presence of agent in lymphoid tissue |
Not readily detected |
Readily detected |
| Increased glycoform ratio on immunoblot
analysis of protease-resistance prion protein |
Not reported |
Marked accumulation of protease-resistance prion
protein |
| Source: Adapted from
Belay E., Schonberger L. Variant Creutzfeldt-Jakob Disease and Bovine
Spongiform Encephalopathy. Clin Lab Med 2002;22:849-62. |
| *An abnormal signal in
the posterior thalami on T2- and diffusion-weighted images and
fluid-attenuated inversion recovery sequences on brain magnetic resonance
imaging (MRI); in the appropriate clinical context, this signal is highly
specific for vCJD. |
Reference in this website to any specific
commercial products, process, service, manufacturer,
or company does not constitute its endorsement or recommendation by
the U.S. Government or CDC. CDC is not responsible for the contents
of any "off-site" web
page referenced from this server.
|
