
| | | | |
Environmental Medicine
|
Amyotrophic Lateral Sclerosis, Lead, and Genetic Susceptibility: Polymorphisms in the 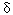 -Aminolevulinic Acid Dehydratase and Vitamin D Receptor Genes -Aminolevulinic Acid Dehydratase and Vitamin D Receptor Genes Freya Kamel,1 David M. Umbach,1 Teresa A. Lehman,2 Lawrence P. Park,3 Theodore L. Munsat,4 Jeremy M. Shefner,5 Dale P. Sandler,1 Howard Hu,6 and Jack A. Taylor1
1National Institute of Environmental Health Sciences, Research Triangle Park, North Carolina, USA; 2Bioserve Biotechnologies, Rockville, Maryland, USA; 3Westat, Durham, North Carolina, USA; 4New England Medical Center, Boston, Massachusetts, USA; 5SUNY Upstate Medical University, Syracuse, New York, USA 6Harvard Medical School and Harvard School of Public Health, Boston, Massachusetts, USA
Abstract
Previous studies have suggested that lead exposure may be associated with increased risk of amyotrophic lateral sclerosis (ALS) . Polymorphisms in the genes for -aminolevulinic acid dehydratase (ALAD) and the vitamin D receptor (VDR) may affect susceptibility to lead exposure. We used data from a case-control study conducted in New England from 1993 to 1996 to evaluate the relationship of ALS to polymorphisms in ALAD and VDR and the effect of these polymorphisms on the association of ALS with lead exposure. The ALAD 2 allele (177G to C ; K59N) was associated with decreased lead levels in both patella and tibia, although not in blood, and with an imprecise increase in ALS risk [odds ratio (OR) = 1.9 ; 95% confidence interval (95% CI) , 0.60-6.3]. We found a previously unreported polymorphism in ALAD at an Msp1 site in intron 2 (IVS2+299G>A) that was associated with decreased bone lead levels and with an imprecise decrease in ALS risk (OR = 0.35 ; 95% CI, 0.10-1.2) . The VDR B allele was not associated with lead levels or ALS risk. Our ability to observe effects of genotype on associations of ALS with occupational exposure to lead or with blood or bone lead levels was limited. These findings suggest that genetic susceptibility conferred by polymorphisms in ALAD may affect ALS risk, possibly through a mechanism related to internal lead exposure. Key words: -aminolevulinic acid dehydratase, amyotrophic lateral sclerosis, genetic susceptibility, lead, vitamin D receptor. Environ Health Perspect 111:1335-1339 (2003) . doi:10.1289/ehp.6109 available via http://dx.doi.org/ [Online 1 April 2003] |
|
|
|
Address correspondence to F. Kamel, Epidemiology Branch, National Institute of Environmental Health Sciences, Box 12233, MD A3-05, Research Triangle Park, NC 27709 USA. Telephone: (919) 541-1581. Fax: (919) 541-2511. E-mail: kamel@niehs.nih.gov
We gratefully acknowledge the work of L. Lansdell and K. Catoe in conducting the case-control study and the generous contributions of time and effort made by the study participants. This work was supported by intramural funding to the Epidemiology Branch, National Institute of Environmental Health Sciences, National Institutes of Health.
The authors declare they have no conflict of interest.
Received 14 November 2002; accepted 1 April 2003.
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease affecting the motor neurons of the brain and spinal cord. The disease is characterized by muscular atrophy and weakness due to degeneration of spinal motor neurons and by hyperreflexia after loss of cerebral cortical motor neurons. Approximately 5-10% of ALS cases have a family history of ALS. The etiology of ALS remains largely unknown, although genetic factors are likely involved in the familial form (Al-Chalabi and Leigh 2000). Environmental exposures have also been considered as potential causes of ALS (Nelson 1995-1996). We reported that increased risk of ALS was associated with occupational exposure to lead and with higher levels of both bone and blood lead, suggesting a potential role for lead exposure in the etiology of ALS (Kamel et al. 2002).
Genetic susceptibility may modify the relationship of ALS to lead exposure. A potentially relevant gene is ALAD, found on chromosome 9q34 (Kelada et al. 2001), which codes for -aminolevulinic acid dehydratase (ALAD), an enzyme involved in heme synthesis in red blood cells. A G to C transversion at position 177 of the coding region of ALAD replaces a lysine with an asparagine at position 59 of the ALAD protein, creating a variant allele ALAD 2 as opposed to the wild-type allele ALAD 1 (Wetmur et al. 1991a). The frequency of ALAD 2 is approximately 10% in Caucasian populations (Kelada et al. 2001). The ALAD enzyme is the principal lead-binding site in erythrocytes, and the ALAD 2 protein binds lead more tightly than does the ALAD 1 protein (Bergdahl et al. 1997b). This change alters the toxicokinetics of lead and may modify risk associated with lead exposure (Kelada et al. 2001).
VDR, another gene potentially affecting susceptibility to lead, is found on chromosome 12q and codes for the vitamin D receptor (VDR). One polymorphism in this gene is found at a BsmI restriction site in the intron separating exons VIII and IX (Zmuda et al. 2000). Presence of the site is denoted by b and absence by B. The BB genotype is found in 10-20% of Caucasians (Cooper and Umbach 1996). The BB genotype may be associated with a less functional receptor, thereby affecting calcium absorption and distribution in the body (Zmuda et al. 2000). Vitamin D can also influence lead absorption and distribution (Fullmer 1992), suggesting that the BB genotype might also be associated with uptake of lead or susceptibility to lead toxicity. Because the polymorphism is located in an intron and does not appear to affect splicing (Zmuda et al. 2000), it is unlikely to cause direct changes in VDR function, but it may be in linkage disequilibrium with functional variants within the VDR gene or in another closely linked gene.
We used data from a case-control study conducted in New England from 1993 to 1996 (Kamel et al. 2002) to investigate associations of ALS with polymorphisms in ALAD and VDR and the influence of genotype on the previously observed association of ALS with lead exposure.
Population. We recruited cases from two major referral centers in New England: the Neuromuscular Research Unit at New England Medical Center and the Neurophysiology Laboratory at Brigham and Women's Hospital (Boston, MA). Diagnosis of ALS was based on criteria published by the World Federation of Neurology (Brooks 1994) and confirmed by a board-certified neurologist (T.L.M. or J.M.S.). Patients were eligible if they had received an initial diagnosis of ALS within the 2 years before enrollment, if they lived in New England at least half the year, and if they spoke English and were mentally competent. Population controls were identified by random telephone screening (Waksberg 1978) and were eligible if they lived in New England at least half the year, spoke English, and were mentally competent. In addition, potential controls were excluded if they had a physician diagnosis of any neurodegenerative disease, neuropathy, or post-polio syndrome. We frequency-matched controls to cases so that the distributions of the variables age, sex, and region within New England were similar in the two groups.
The institutional review boards of the National Institute of Environmental Health Sciences, New England Medical Center, Brigham and Women's Hospital, Survey Research Associates-Battelle (Durham, NC), and CODA (Durham, NC) approved the study. All participants gave informed consent.
Questionnaire. We collected information on demographic and lifestyle characteristics using a structured interview administered by trained personnel (Kamel et al. 2002). Of those eligible, 71% of cases and 76% of controls completed the interview. Information on diet 5 years before interview was collected with a food frequency questionnaire (Longnecker et al. 2000). Interview-based variables considered in the present study were age in years (continuous variable), sex, region (outside vs. within Boston city limits), education (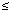 high school vs. > high school), current physical activity (hours per day spent sitting, lying down, or sleeping; continuous variable), cigarette smoking (ever smoked at least 100 cigarettes vs. never smoked), alcohol use (ever had at least 10 drinks of beer, wine, or liquor vs. never), occupational exposure to lead (ever had a job that involved exposure to lead fumes, dust, or particles 10 or more times vs. never), and daily calcium intake in grams (continuous variable), based on both food and supplements. high school vs. > high school), current physical activity (hours per day spent sitting, lying down, or sleeping; continuous variable), cigarette smoking (ever smoked at least 100 cigarettes vs. never smoked), alcohol use (ever had at least 10 drinks of beer, wine, or liquor vs. never), occupational exposure to lead (ever had a job that involved exposure to lead fumes, dust, or particles 10 or more times vs. never), and daily calcium intake in grams (continuous variable), based on both food and supplements.
Measurement of blood and bone lead. We invited all cases and a subset of the controls who lived within 20 miles of the testing center to come to the laboratory for collection of blood samples and measurement of bone lead. Response rates for this portion of the study were 95% for cases and 41% for controls. Controls who were invited but declined to come in to the laboratory were similar in age, sex, education, physical activity, smoking, and alcohol use to those who participated (Kamel et al. 2002). Blood lead (micrograms per deciliter) was measured using graphite furnace atomic absorption spectroscopy. Bone lead was measured in the mid-tibial shaft and the patella using in vivo K X-ray fluorescence (K-XRF) as previously described (Aro et al. 1994; Burger et al. 1990; Kamel et al. 2002). The technique provides an unbiased estimate of bone lead levels as micrograms of lead per gram of bone mineral. Negative estimates of bone lead concentration may be obtained when true values are close to zero.
DNA isolation and genotyping. Genomic DNA was isolated from ~9 mL of frozen whole blood using Gentra PUREGENE reagents (Gentra Systems, Minneapolis, MN). The region surrounding the known ALAD polymorphic site was amplified using a slight modification of a published method (Hsieh et al. 2000) with identical primers. Amplification reactions were performed in 50 µL total volume with 100 ng genomic DNA, 20 pmol of each primer, 1 Qiagen polymerase chain reaction (PCR) buffer containing Mg2+ (Qiagen, Valencia, CA), 37.5 µM of each dNTP, and 2.5 U Taq polymerase (Life Technologies, Carlsbad, CA). The cycling conditions were 94°C for 5 min followed by 35 cycles of 94°C for 1 min, 56°C for 1 min, and 72°C for 1 min, with a final cycle at 72°C for 7 min. A 10-µL aliquot of each PCR solution was digested with 20 U (2 µL) MspI restriction enzyme (New England Biolabs, Beverly, MA) in 2.5 µL 10 NEBuffer 2 (50 mM NaCl, 10 mM Tris-HCl, 1 mM dithiothreitol) with 10.5 µL water at 37°C for 3 hr. The digested fragments were separated on a 2% agarose gel. Diagnostic fragment sizes were 582 bp for ALAD 1 and 511 bp + 71 bp for ALAD 2. We also identified a novel polymorphism in intron 2 of ALAD. Qiagen polymerase chain reaction (PCR) buffer containing Mg2+ (Qiagen, Valencia, CA), 37.5 µM of each dNTP, and 2.5 U Taq polymerase (Life Technologies, Carlsbad, CA). The cycling conditions were 94°C for 5 min followed by 35 cycles of 94°C for 1 min, 56°C for 1 min, and 72°C for 1 min, with a final cycle at 72°C for 7 min. A 10-µL aliquot of each PCR solution was digested with 20 U (2 µL) MspI restriction enzyme (New England Biolabs, Beverly, MA) in 2.5 µL 10 NEBuffer 2 (50 mM NaCl, 10 mM Tris-HCl, 1 mM dithiothreitol) with 10.5 µL water at 37°C for 3 hr. The digested fragments were separated on a 2% agarose gel. Diagnostic fragment sizes were 582 bp for ALAD 1 and 511 bp + 71 bp for ALAD 2. We also identified a novel polymorphism in intron 2 of ALAD.
The region surrounding the known VDR polymorphic site was amplified using previously described primers (Morrison et al. 1994). Amplification reactions were performed in 35 µL total volume with 100 ng genomic DNA, 10 pmol each of primer, 1 BRL PCR buffer containing 1.75 mM Mg2+ (Life Technologies), 37.5 µM each dNTP, and 2 U Taq polymerase (Life Technologies). The cycling conditions were 94°C for 5 min followed by 35 cycles of 94°C for 1 min, 60°C for 1 min, and 72°C for 1 min, with a final cycle at 72°C for 7 min. A 11.5 µL aliquot of each PCR was digested with 20 U (2 µL) BsmI restriction enzyme (New England Biolabs) in 1.5 µL 10 NEBuffer 2 with 2 µL water at 37°C for 3 hr. The digested fragments were separated on a 1% agarose gel. Diagnostic fragment sizes were 800 bp for B and 650 bp + 150 bp for b.
|
Table 1.

|
Data analysis. The present analysis, conducted using SAS (version 8.2; SAS Institute, Inc., Cary, NC), includes 103 cases and 38 controls who provided complete data. Lead levels were modeled as continuous variables to increase the statistical power of the analysis. Blood lead levels below assay sensitivity were assigned a value of 0.5 µg/dL, one-half the detection limit. To remove extreme skewness in bone lead distribution while accommodating the few negative values, we transformed bone lead concentrations using the function log2 (Pb + 32), where Pb is bone lead concentration in micrograms per gram (Kamel et al. 2002). Because few homozygotes were found for either polymorphism in ALAD (Table 1), ALAD genotypes were included as dichotomous predictors, indicating the presence or absence of the polymorphism. No evidence of dose response was observed in models comparing VDR bB or VDR BB to VDR bb, so results are presented from models using dichotomous predictors.
The relationship of genotype to blood or bone lead levels was analyzed in the control group alone because inclusion of cases might have allowed disease to distort the relationship. We used linear regression for these analyses, with blood lead, log-transformed patella lead, or log-transformed tibia lead as the dependent variable. On the basis of preliminary analyses, we considered the following independent variables: age, square root of age, sex, region, education, inactivity, cigarette smoking, occupational exposure to lead, and patella lead (for blood lead models only). We used backward elimination to determine which of these covariates to include in base models, using p = 0.15 as a cutoff. The base model for blood lead included variables for age, sex, region, inactivity, cigarette smoking, and patella lead; the base model for patella lead included age and cigarette smoking; and the base model for tibia lead included age, sex, and region. Genotype variables were added to these base models. Results of linear regressions are presented as estimated coefficients with 95% confidence intervals (95% CIs) based on standard errors.
We analyzed the relationship of ALS to lead exposure or genotype using multiple logistic regression. All models included the matching variables age, sex, and region. Both education and current physical activity levels were associated with case-control status and also potentially related to blood and bone lead levels, so models also included these variables. VDR models were also constructed with or without calcium intake. Associations are estimated as odds ratios (ORs), and 95% CIs are based on standard errors. No substantive differences were found when familial cases were excluded from the analyses, so results for all participants are presented.
We used standard methods (Weir 1996) to evaluate Hardy-Weinberg equilibrium at each locus and to examine linkage disequilibrium between the two polymorphisms in ALAD.
Blood lead levels in study participants ranged from < 1 to 14 µg/dL; patella lead levels from -4 to 107 µg/g; and tibia lead levels from -7 to 61 µg/g (Kamel et al. 2002). Thirty-six percent of cases and 21% of controls reported ever having had a job involving lead exposure. Ninety-six percent of cases and 92% of controls were white and not Hispanic.
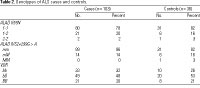
We found a novel polymorphism in ALAD at an MspI site in intron 2; a G to A transition at nucleotide 299 destroyed the site (GeneSNPs 2003). Diagnostic fragment sizes for ALAD IVS2+299G>A were 160 bp and 138 bp for ALAD I2-1 and 298 bp for ALAD I2-2. We confirmed the presence of the polymorphism by sequencing (data not shown); it was not located at a splice site. We denote the polymorphism at position 177 by ALAD K59N, and, following convention, its alleles by ALAD 1 (wild type) and ALAD 2 (variant). We denote the intron 2 polymorphism by ALAD IVS2+299G>A and its alleles by ALAD I2-1 (wild type) and ALAD I2-2 (variant). The relative distribution of the two ALAD polymorphisms is shown in Table 1. We saw no evidence of linkage disequilibrium between ALAD K59N and ALAD IVS2+299G > A in either cases (p = 0.3) or controls (p = 0.5).
|
Table 2.
|
|
Table 3.

|
|
Table 4.
|
|
Table 5.

|
ALAD K59N genotype. We found no deviation from Hardy-Weinberg equilibrium for the two ALAD K59N alleles in either cases (p = 0.7) or controls (p = 0.3). The genotype frequencies are shown in Table 2. The frequency of the ALAD 2 allele was 12% in cases and 11% in controls; the crude OR for the relationship of the variant allele to ALS was 1.3 (95% CI, 0.5-3.7). In controls, presence of the allele was associated with decreases in patella and tibia lead levels but not with blood lead levels (Table 3). The ALAD 2 allele was not related to the distribution of lead among the three lead compartments (data not shown).
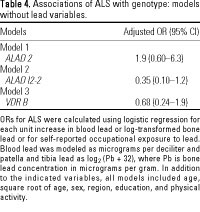
After adjustment for age, sex, region, education, and physical activity, ALAD 2 was associated with an approximately 2-fold increase in risk of ALS, although the relationship was imprecise (Table 4, model 1). The association of ALAD 2 with ALS risk was strengthened by further adjustment for blood lead, although adjustment for patella or tibia lead or occupational exposure to lead made little difference (Table 5, model 5). ORs for blood and bone lead and occupational lead exposure were unchanged by adjustment for ALAD 2 (Table 5, models 4 and 5). We saw no important interaction of ALAD 2 with any lead variable (data not shown). Results from models that were not adjusted for physical activity (data not shown) were similar to those presented in Tables 4 and 5.
ALAD IVS2+299G>A genotype. We found no deviation from Hardy-Weinberg equilibrium for the two ALAD IVS2+299G>A alleles in either cases (p = 0.5) or controls (p = 0.3). The genotype frequency is given in Table 2. The frequency of the ALAD I2-2 allele was 7% in cases and 11% in controls; the crude OR for the relationship of ALS to the presence of the variant allele was 0.7 (95% CI, 0.2-2.1). In controls, presence of the ALAD I2-2 allele was associated with decreases in patella and tibia lead levels but not with blood lead levels (Table 3).
After adjustment for age, sex, region, education, and physical activity, presence of the ALAD I2-2 allele was inversely associated with ALS, with or without adjustment for lead levels or occupational exposure to lead (Table 4, model 2; Table 5, model 6). ORs for blood and bone lead and occupational lead exposure were unchanged by adjustment for ALAD I2-2 (Table 5, models 4 and 6). We saw a large but imprecise interaction between ALAD I2-2 and lead variables that was likely due to small numbers (data not shown). Results from models that were not adjusted for physical activity (data not shown) were similar to those presented in Tables 4 and 5. In models including both ALAD 2 and ALAD I2-2, ORs for both polymorphisms were similar to those found in models with only one genotype, whether or not lead variables were also included (data not shown).
VDR genotype. We found no deviation from Hardy-Weinberg equilibrium for the two VDR alleles in either cases or controls (p = 0.7 for each). The genotype frequency is given in Table 2. The frequency of the B allele was 45% in cases and 48% in controls; the crude OR for the presence of the variant allele was 0.8 (95% CI, 0.3-1.9). Presence of the allele was not associated with either blood or bone lead levels in controls (Table 3).
After adjustment for age, sex, region, education, and physical activity, VDR B was not related to ALS, without or with adjustment for lead levels or occupational exposure to lead (Table 4, model 3; Table 5, model 7). ORs for blood and bone lead and occupational lead exposure were unchanged by adjustment for VDR B (Table 5, models 4 and 7). Further adjustment for calcium intake did not alter these relationships (data not shown). We saw no important interaction of VDR B with any lead variable (data not shown). Results from models that were not adjusted for physical activity (data not shown) were similar to those presented in Tables 4 and 5. Results from models comparing VDR bB or VDR BB with VDR bb were similar to results using the dichotomous variable; we found no evidence of a dose response (data not shown).
In this study, we found that the ALAD 2 allele (177G to C; K59N) and a previously unidentified polymorphism at an MspI site in intron 2 of ALAD, denoted ALAD I2-2, were both associated with decreased bone lead levels but not with blood lead levels. ALAD 2 appeared to be associated with an increase and ALAD I2-2 with a decrease in risk of ALS, although both associations were imprecise. In contrast, the VDR B allele showed no relationship to blood or bone lead levels or to ALS risk.
Our finding that ALAD 2 was associated with decreases in both tibia and patella lead levels but not with blood lead levels is consistent with some previous results. Some studies have reported that blood lead levels were elevated in ALAD 2 carriers, although the differences were not always statistically significant (Alexander et al. 1998; Fleming et al. 1998; Hsieh et al. 2000; Schwartz et al. 2000a; Wetmur et al. 1991b; Ziemsen et al. 1986). Other studies have found no difference in blood lead levels between ALAD 1 homozygotes and ALAD 2 carriers (Bergdahl et al. 1997a; Hu et al. 2001; Lee BK et al. 2001; Schwartz et al. 1997; Smith et al. 1995a). Any difference between genotypes in blood lead levels is likely due to tighter binding of lead to ALAD 2 than to ALAD 1 (Bergdahl et al. 1997b) and may be evident only at higher blood lead concentrations where other binding sites are saturated (Hu et al. 2001). Thus, a difference would not necessarily be expected at the relatively low blood lead concentrations found in our study participants. ALAD 2 has also been associated with decreased lead levels in trabecular or cortical bone (Hu et al. 2001; Smith et al. 1995b) and with decreased uptake of lead into both bone compartments (Fleming et al. 1998), although other studies found no relationship (Bergdahl et al. 1997a; Lee BK et al. 2001; Schwartz et al. 2000a).
Overall these findings are consistent with the hypothesis that ALAD 2 alters the toxicokinetics of lead, promoting retention of lead in blood and migration of lead from bone to blood. The implications for lead toxicity are unclear. Tighter binding to ALAD 2 in red blood cells could make lead less available to target tissues and hence less toxic. On the other hand, increased retention of lead in blood relative to bone might increase its availability to target tissues. Our data suggesting that ALAD 2 was positively associated with ALS are consistent with the latter hypothesis.
The effect of the ALAD 2 allele on lead toxicokinetics might in theory modify associations of ALS with indices of internal exposure (blood and bone lead) or external exposure (occupational exposure). We found no evidence to support either of these possibilities. The ALAD 2 polymorphism did not alter the relationship of blood or bone lead to ALS or the risk associated with occupational exposure to lead. However, our study had limited power to evaluate effect modification, and this issue needs further consideration.
Few previous studies have examined the effect of ALAD 2 on health outcomes. ALAD 2 had no consistent relationship to hematologic parameters (Alexander et al. 1998; Lee SS et al. 2001; Schwartz et al. 1995; Sithisarankul et al. 1997; Smith et al. 1995a) or to blood pressure or hypertension (Lee BK et al. 2001; Smith et al. 1995b). One study found detrimental effects of ALAD 2 on renal function (Smith et al. 1995b) and another found better performance on a test of attention in five carriers of the allele (Bellinger et al. 1994). A recent study found that ALAD 2 modified the association of bone lead levels with indices of renal function (Wu et al. 2003), but other studies have not detected effect modification (Alexander et al. 1998; Lee BK et al. 2001; Lee SS et al. 2001).
We found no association of VDR B with blood or bone lead levels or with ALS. In several studies of a group of Korean lead workers, Schwartz and colleagues found that VDR B was associated with an increase in blood and tibia lead levels and with increased blood pressure and hypertension, although not with hematopoietic outcomes (Lee BK et al. 2001; Lee SS et al. 2001; Schwartz et al. 2000a). VDR B did not modify the effect of lead on blood pressure and hypertension (Lee BK et al. 2001) but was weakly associated with a decreased effect of lead on hemoglobin and hematocrit (Lee SS et al. 2001). In another cohort, VDR B had only minimal effects on tibia lead levels but increased the accumulation of tibia lead with age (Schwartz et al. 2000b). Understanding the relationship of this polymorphism to lead toxicokinetics thus awaits further study.
It is unclear why previous studies of ALAD 2 have not reported the MspI polymorphism in intron 2. The 2% agarose gels we used to separate the reaction products may provide better resolution of the shorter fragments associated with this polymorphism. Nor is it clear why ALAD IVS2+299G>A, an intronic variant, is associated with changes in bone lead levels or with ALS. The associations do not appear to be the result of strong linkage or other interaction with the ALAD K59N site. Recent work has demonstrated that intronic mutations can have functional consequences in some genes--for example, p53 (Lehman et al. 2000).
Our study is limited by the low participation rate of controls in the laboratory portion of the study. This problem raises concerns about selection bias and also reduces the power of the study, contributing to the imprecision of some estimates. However, none of the three genotypes was seen to deviate from Hardy-Weinberg equilibrium, and frequencies of ALAD 2 and VDR B were similar to reported values (Cooper and Umbach 1996; Kelada et al. 2001). Further, controls who were invited but declined to participate in the lab portion of the study were similar in all characteristics examined to those who participated (Kamel et al. 2002). We therefore regard it as unlikely that the association of ALS with ALAD genotype can be explained entirely by selection bias.
Previously we reported that ALS risk was associated with increases in both blood and bone lead levels (Kamel et al. 2002). The association with blood lead was surprisingly strong, with a 2-fold increase in risk for each microgram per decaliter increase in blood lead levels. Although selection bias might have influenced our results, some of the potential biases, if present, would likely have minimized the association of ALS with blood lead rather than creating a spurious relationship (Kamel et al. 2002). Blood lead is often considered to reflect recent exposure. However, in adults without current exogenous exposure, like most of the participants in our study, bone lead is the major source of blood lead (Hu et al. 1998). The latter may therefore reflect cumulative lifetime exposure. Blood lead levels are determined by bone lead levels together with factors affecting mobilization of lead from bone. The present study suggests that ALAD genotype may be one such factor, whereas VDR genotype does not appear to be important. Alterations in lead toxicokinetics conferred by the presence of the ALAD 2 allele may subtly increase exposure to lead throughout a person's lifetime, thereby elevating risk. We cannot exclude the possibility that ALAD 2 affects ALS risk through some mechanism independent of lead exposure, perhaps through an unidentified polymorphism in linkage disequilibrium with the ALAD 2 site. However, the association of ALAD 2 with decreased bone lead levels suggests that lead may play some role.
In conclusion, our study suggests that genetic susceptibility conferred by ALAD 2 is associated with ALS risk, possibly through a mechanism related to lead exposure. Because our study is small, and the observation is unique, this hypothesis needs further consideration.
|
|
|
| [References Listed in PubMed]
References
Al-Chalabi A, Leigh PN. 2000. Recent advances in amyotrophic lateral sclerosis. Curr Opin Neurol 13(4):397-405.
Alexander BH, Checkowy H, Costa-Mallen P, Faustman EM, Woods JS, Kelsey KT, et al. 1998. Interaction of blood lead and delta-aminolevulinic acid dehydratase genotype on markers of heme synthesis and sperm production in lead smelter workers. Environ Health Perspect 106:213-216.
Aro ACA, Todd AC, Amarasiriwardena C, Hu H. 1994. Improvements in the calibration of CD-109 K x-ray fluorescence systems for measuring bone lead in vivo. Phys Med Biol 39(12):2263-2271.
Bellinger D, Hu H, Titlebaum L, Needleman HL. 1994. Attentional correlates of dentin and bone lead levels in adolescents. Arch Environ Health 49(2):98-105.
Bergdahl IA, Gerhardsson L, Schutz A, Desnick RJ, Wetmur JG, Skerfving S. 1997a. Delta-aminolevulinic acid dehydratase polymorphism: influence on lead levels and kidney function in humans. Arch Environ Health 52(2):91-96.
Bergdahl IA, Grubb A, Schutz A, Desnick RJ, Wetmur JG, Sassa S, et al. 1997b. Lead binding to delta-aminolevulinic acid dehydratase (ALAD) in human erythrocytes. Pharmacol Toxicol 81(4):153-158.
Brooks BR. 1994. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. J Neurol Sci 124(suppl):96-107.
Burger DE, Milder FL, Morsillo PR, Adams BB, Hu H. 1990. Automated bone lead analysis by K-x-ray fluorescence for the clinical environment. Basic Life Sci 55:287-292.
Cooper GS, Umbach DM. 1996. Are vitamin D receptor polymorphisms associated with bone mineral density? A meta-analysis. J Bone Min Res 11(12):1841-1849.
Fleming DEB, Chettle DR, Wetmur JG, Desnick RJ, Robin JP, Bouleay D, et al. 1998. Effect of the delta-aminolevulinate dehydratase polymorphism of the accumulation of lead in bone and blood in lead smelter workers. Environ Res A77:49-61.
Fullmer CS. 1992. Intestinal interactions of lead and calcium. Neurotoxicology 13(4):799-807.
Hsieh LL, Liou SH, Chen YH, Tsai LC, Yang T, Wu TN. 2000. Association between aminolevulinate dehydrogenase genotype and blood lead levels in Taiwan. J Occup Environ Med 42(2):151-155.
Hu H, Rabinowitz M, Smith D. 1998. Bone lead as a biological marker in epidemiologic studies of chronic toxicity: conceptual paradigms. Environ Health Perspect 106:1-8.
Hu H, Wu MT, Cheng Y, Sparrow D, Weiss S, Kelsey K. 2001. The delta-aminolevulinic acid dehydratase (ALAD) polymorphism and bone and blood lead levels in community-exposed men: the Normative Aging Study. Environ Health Perspect 109:827-832.
Kamel F, Umbach DM, Munsat TL, Shefner JM, Hu H, Sandler DP. 2002. Lead exposure and amyotrophic lateral sclerosis. Epidemiology 13(3):311-319.
Kelada SN, Shelton E, Kaufmann RB, Khoury MJ. 2001. Delta-aminolevulinic acid dehydratase genotype and lead toxicity: a HuGE review. Am J Epidemiol 154(1):1-13.
Lee BK, Lee GS, Stewart WF, Ahn KD, Simon D, Kelsey KT, et al. 2001. Associations of blood pressure and hypertension with lead dose measures and polymorphisms in the vitamin D receptor and delta-aminolevulinic acid dehydratase genes. Environ Health Perspect 109:383-389.
Lee SS, Lee BK, Lee GS, Stewart WF, Simon D, Kelsey K, et al. 2001. Associations of lead biomarkers and delta-aminolevulinic acid dehydratase and vitamin D receptor genotypes with hematopoietic outcomes in Korean lead workers. Scand J Work Environ Health 27(6):402-411.
Lehman TA, Haffty BG, Carbone CJ, Bishop LR, Gumbs AA, Krishnan S, et al. 2000. Elevated frequency and functional activity of a specific germ-line p53 intron mutation in familial breast cancer. Cancer Res 60:1062-1069.
Longnecker MP, Kamel F, Umbach DM, Munsat TL, Shefner JM, Lansdell LW, et al. 2000. Dietary intake of calcium, magnesium and antioxidants in relation to risk of amyotrophic lateral sclerosis. Neuroepidemiology 19(4):210-216.
Morrison NA, Qi JC, Tokita A, Kelly PJ, Crofts L, Nguyen TV, et al. 1994. Prediction of bone density from vitamin D receptor alleles. Nature 367(6460):284-287.
Nelson LM. 1995-1996. Epidemiology of ALS. Clin Neurosci 3(6):327-331.
NIEHS. 2003. GeneSNPs. Research Triangle Park, NC:National Institute of Environmental Health Sciences. Available: http://www.genome.utah.edu/genesnps/ [accessed 9 June 2003].
Schwartz BS, Lee BK, Lee GS, Stewart WF, Simon D, Kelsey K, et al. 2000a. Associations of blood lead, dimercaptosuccinic acid-chelatable lead, and tibia lead with polymorphisms in the vitamin D receptor and [delta]-aminolevulinic acid dehydratase genes. Environ Health Perspect 108:949-954.
Schwartz BS, Lee BK, Stewart W, Ahn KD, Springer K, Kelsey K. 1995. Associations of delta-aminolevulinic acid dehydratase genotype with plant, exposure duration, and blood lead and zinc protoporphyrin levels in Korean lead workers. Am J Epidemiol 142(7):738-745.
Schwartz BS, Lee BK, Stewart W, Sithisarankul P, Strickland PT, Ahn KD, et al. 1997. delta-Aminolevulinic acid dehydratase genotype modifies four hour urinary lead excretion after oral administration of dimercaptosuccinic acid. Occup Environ Med 54(4):241-246.
Schwartz BS, Stewart WF, Kelsey KT, Simon D, Park S, Links JM, et al. 2000b. Associations of tibial lead levels with BsmI polymorphisms in the vitamin D receptor in former organolead manufacturing workers. Environ Health Perspect 108:199-203.
Sithisarankul P, Schwartz BS, Lee BK, Kelsey KT, Strickland PT. 1997. Aminolevulinic acid dehydratase genotype mediates plasma levels of the neurotoxin, 5-aminolevulinic acid, in lead-exposed workers. Am J Ind Med 32(1):15-20.
Smith CM, Hu H, Wang X, Kelsey KT. 1995a. ALA-D genotype is not associated with HT or HB levels among workers exposed to low levels of lead. Med Lav 86(3):229-235.
Smith CM, Wang X, Hu H, Kelsey KT. 1995b. A polymorphism in the delta-aminolevulinic acid dehydratase gene may modify the pharmacokinetics and toxicity of lead. Environ Health Perspect 103:248-253.
Waksberg J. 1978. Sampling methods for random digit dialing. J Am Statis Assoc 73(361):40-46.
Weir BS. 1996. Genetic Data Analysis II. Sunderland, MA:Sinauer Associates, Inc.
Wetmur JG, Kaya AH, Plewinska M, Desnick RJ. 1991a. Molecular characterization of the human -aminolevulinate dehydratase 2 (ALAD2) allele: implications for molecular screening of individuals for genetic susceptibility to lead poisoning. Am J Hum Genet 49(4):757-763.
Wetmur JG, Lehnert G, Desnick RJ. 1991b. The -aminolevulinate dehydratase polymorphism: higher blood lead levels in lead workers and environmentally exposed children with the 1-2 and 2-2 isozymes. Environ Res 56:109-119.
Wu MT, Kelsey K, Schwartz J, Sparrow D, Weiss S, Hu H. 2003. A -aminolevulinic acid deydratase (ALAD) polymorphism may modify the relationship of low-level lead exposure to uricemia and renal function: the normative aging study. Environ Health Perspect 111:335-340.
Ziemsen B, Angerer J, Lehnert G, Benkmann HG, Goedde HW. 1986. Polymorphism of delta-aminolevulinic acid dehydratase in lead-exposed workers. Int Arch Occup Environ Health 58:245-247.
Zmuda JM, Cauley JA, Ferrell RE. 2000. Molecular epidemiology of vitamin D receptor gene variants. Epidemiol Rev 22(2):203-217.
Last Updated: July 10, 2003 |
|
|
|
| |









