


Guidance for Industry
Biological Product Deviation Reporting for Blood and Plasma Establishments
Additional copies of this guidance document are available from the Office of Communication, Training and Manufacturers Assistance, (HFM-40), 1401 Rockville Pike, Suite 200N, Rockville, MD 20852-1448, or by calling 1-800-835-4709 or 301-827-1800, or from the Internet at http://www.fda.gov/cber/guidelines.htm.
For questions on the content of this guidance, contact (CBER) Office of Compliance and Biologics Quality at 301-827-6220 or by email at bp_deviations@fda.hhs.gov.
U.S. Department of Health and Human Services
Food and Drug Administration
Center for Biologics Evaluation and Research
October 2006
- INTRODUCTION
- BACKGROUND
- GUIDANCE
- Who Must Report? (21 CFR 606.171(a))
Examples of who must report: - What Do I Report? (21 CFR 606.171(b))
Biological Product Deviation Reporting Flow Chart for Blood and Plasma Establishments - When Do I Report? (21 CFR 606.171(c))
- How and Where Do I Report? (21 CFR 606.171(d) and (e))
- EXAMPLES OF REPORTABLE AND NON-REPORTABLE EVENTS BY MANUFACTURING SYSTEM
- Donor Suitability
- Post Donation Information
- Donor Screening and Deferral
- Collection
- Component Preparation
- Testing
- Labeling
- Quality Control and Distribution
- Miscellaneous
- REFERENCES
Contains Nonbinding Recommendations
Guidance for Industry
Biological Product Deviation Reporting for Blood and Plasma Establishments
| This guidance represents the Food and Drug Administration's (FDA's) current thinking on this topic. It does not create or confer any rights for or on any person and does not operate to bind FDA or the public. You can use an alternative approach if the approach satisfies the requirements of the applicable statutes and regulations. If you want to discuss an alternative approach, contact the appropriate FDA staff. If you cannot identify the appropriate FDA staff, call the appropriate number listed on the title page of this guidance. |
- INTRODUCTION
- BACKGROUND
- Expands the reporting requirement to include all unlicensed blood establishments, including transfusion services and registered blood banks;
- Replaces the term "error and accident" with "biological product deviation";
- More clearly describes the types of events that you must report to us;
- Limits reporting to those events that may affect the safety, purity, or potency of distributed products;
- Establishes a reporting time frame of 45 days from the date the event was discovered.
- a timely investigation;
- an appropriate corrective action plan to prevent recurrence;
- procedures to gain control of unsuitable products in a timely manner;
- appropriate disposition of all affected products (in-date and expired);
- for deviations and discrepancies associated with donor suitability, an assessment of the donor's suitability to serve as a donor in the future.
- GUIDANCE
- Who Must Report? (21 CFR 606.171(a))
- licensed manufacturers of blood and blood components, including Source Plasma;
- unlicensed registered blood establishments; and
- transfusion services.
- EVENT
You are a blood establishment that contracts with a test laboratory to have the laboratory perform viral marker testing, including nucleic acid testing (NAT). The test laboratory did not perform the testing in accordance with the test kit manufacturer's instructions or its own procedures. The test laboratory used the incorrect incubation time. Failure to follow the test kit instructions or validated procedures may affect the safety, purity, or potency of the product. - EVENT
You are a blood establishment that contracted with another blood establishment (referred to as an irradiator) to perform irradiation of blood products. You sent a product to the irradiator. After irradiation, the product was returned to you. The irradiator notified you that the incorrect dosage was used (i.e., established specifications were not met), which may affect the safety, purity, or potency of the product. - EVENT
You are a blood establishment that distributed a blood product to another blood establishment (referred to as an irradiator), which subsequently irradiated it. The irradiator used the incorrect dosage during irradiation (i.e., established specifications were not met), which may affect the safety, purity, or potency of the product. - EVENT
You are a transfusion service that received a unit of Platelets from a blood establishment. When you received the product, you discovered that the product was labeled with an expiration date of more than the required 5 days (i.e., improperly extended), which may affect the safety, purity, or potency of the product. You notified the blood establishment. - EVENT
You are a transfusion service that received 10 units of Platelets and subsequently pooled them into one unit. You improperly labeled the pooled unit with an incorrectly extended expiration date, which may affect the safety, purity, or potency of the product. - EVENT
You are a transfusion service that received a unit of Red Blood Cells from a blood establishment. The expiration date on the unit was incorrectly extended. According to your Standard Operating Procedures (SOPs), you should have recognized that the expiration was extended beyond any date appropriate for Red Blood Cells. You crossmatched the unit for a patient and distributed it to the surgical floor for transfusion. The surgical floor questioned the expiration date of the unit. You discovered that the unit was labeled with an extended expiration date, which may affect the safety, purity, or potency of the product. - EVENT
You are a Source Plasma establishment that collected and tested a unit of Source Plasma, and shipped it to a fractionator, a licensed manufacturer. You discovered that the viral marker testing was incorrectly performed and subsequently retested a reserve sample of the unit. The unit retested as positive for a viral marker. The safety, purity, or potency of the Source Plasma may be affected. - What Do I Report? (21 CFR 606.171(b))
- Either
- Represents a deviation from current good manufacturing practice, applicable regulations, applicable standards, or established specifications that may affect the safety, purity, or potency of that product; or
- Represents an unexpected or unforeseeable event that may affect the safety, purity, or potency of that product; and
- Occurs in your facility or a facility under contract with you; and
- Involves a distributed blood or blood component.
- When you did not distribute potentially affected products, regardless of the event.
- When you determined, prior to distributing the product, that the event did not actually affect the safety, purity, or potency of the product.
- When you detected the event and prior to distribution, made the appropriate corrections.
- When the event was related to donor safety only and did not have the potential to affect the safety, purity, or potency of the product.
- If your report would simply state that you were late in reporting the event to us.
- A Transfusion Service is NOT required to report to us if the event occurs during transfusion or administration procedures, after the blood product has left the control of the transfusion service. For example, a report is not required:
- If the transfusion service issued a product from the laboratory to the nursing floor, operating room, emergency room, etc., for transfusion and the product was not held at the appropriate temperature outside of the transfusion service prior to transfusion. However, the transfusion service is required to report if the product was returned and the transfusion service determined it to be unsuitable, but contrary to CGMP, applicable regulations, applicable standards, or established specifications, reissued the product.
- If the hospital staff, outside of the blood bank, transfused the wrong patient or transfused a patient with the wrong unit, provided the unit was labeled appropriately and the transfusion service conducted compatibility testing properly. If a complication of a transfusion was confirmed to be fatal, the facility that performed the compatibility testing must submit a fatality report to us in accordance with 21 CFR 606.170(b).
- If the transfusion service issued a filter with the product and the hospital staff did not use the filter at the bedside.
- If the patient has a transfusion reaction that is not related to an event in manufacturing. Fatalities due to Transfusion Related Acute Lung Injury (TRALI) or other transfusion complications must be reported to CBER in accordance with 21 CFR 606.170(b). We also accept voluntary reports of non-fatal TRALI as a serious adverse reaction to transfusions. Such voluntary reports can be submitted via MedWatch by phone at 1-800-FDA-1088, by fax at 1-800-FDA-0178, by US mail at MedWatch, HF-2, 5600 Fishers Lane, Rockville, MD 20852, or by email at http://www.fda.gov/medwatch. For additional information, see the "Dear Colleague" letter published October 19, 2001 at http://www.fda.gov/cber/ltr/trali101901.htm.
- A Blood Establishment is NOT required to report if the event is not associated with its manufacturing process and did not occur while the product was under the control of the blood establishment. For example the blood establishment is not required to report to us:
- If the blood establishment shipped a product to a transfusion service and the transfusion service stored the product at the incorrect temperature. Under 21 CFR 606.171(b), if the transfusion service further distributed the product, the transfusion service must report to us.
- If the blood establishment received an unacceptable product (for example, a hemolyzed unit) from another blood establishment, identified the discrepancy and returned or discarded the product. Under 21 CFR 606.171(b), the blood establishment that distributed the unacceptable product, but not the establishment that received and returned or discarded it, must report to us.
- Post Donation Information – A donor donated blood on one occasion. You followed all donor screening and deferral procedures, and determined that the donor was suitable. At a second donation, the donor disclosed additional information that would have resulted in deferral had the donor disclosed it at the earlier donation, such as information that the donor used intravenous drugs prior to the first donation.
- After you distributed a product, you discovered that you performed compatibility testing using a patient sample that was collected from the wrong patient or labeled with incorrect patient information.
- After you distributed a product, your materials vendor informed you that materials used in the collection or processing of blood and blood components, such as reagents, soft goods, software, or collection device, did not meet all requirements or specifications, and you could not have detected the deviation during your routine incoming material qualification procedures.
- maintaining the continued safety, purity, and potency of the product,
- compliance with applicable product and establishment standards, and
- compliance with current good manufacturing practice.
- If a blood establishment labeled a product with an incorrectly extended expiration date and shipped it to a hospital, under 21 CFR 606.171(b) the blood establishment must report to us.
- If a hospital blood bank received 10 units of platelets from a blood establishment, pooled the platelets, mislabeled the final pooled product with an incorrectly extended expiration date, and issued the product to the nursing staff for transfusion, under 21 CFR 606.171(b) the hospital blood bank must report to us. However, if the blood bank detected and corrected the expiration date before distributing the product, under 21 CFR 606.160(b)(7)(iii), 606.100(c), 211.192, and 211.198, the blood bank must document and investigate the deviation but is not required to report to us.
- When Do I Report? (21 CFR 606.171(c))
- How and Where Do I Report? (21 CFR 606.171(d) and (e))
- EXAMPLES OF REPORTABLE AND NON-REPORTABLE EVENTS BY MANUFACTURING SYSTEM
- A donor suitability event occurs in the donor suitability process ( for example , donor screening, donor deferral procedures and receipt of post donation information) (See Section IV. A).
- A collection event occurs during the collection process (See Section IV. B).
- A component preparation event occurs during the component preparation process (See Section IV. C).
- A testing event occurs during the testing process and includes sample deviations and unexpected events (See Section IV. D).
- A labeling event occurs during the labeling process, which includes identifying the information to include on the label, printing the label, and applying the label to the product (See Section IV. E).
- A quality control and distribution event involves a failure in either the quality control (QC) or distribution systems. This category includes the distribution of a product that did not meet specifications (See Section IV. F).
- Donor Suitability
- Post Donation Information
- Subsequent to the donation, a donor (or third party) provides disqualifying information that would have resulted in donor deferral at the time of donation. The relevant questions were asked at the donation, but the donor did not provide the information. Examples include the following:
- Donor had a history of high risk behavior for hepatitis or HIV (Human Immunodeficiency Virus), such as intravenous (IV) drug use, male to male sex, received an accidental needlestick, or transfusion within the time frame for deferral according to your SOP, and did not report this at the time of donation.
- Donor traveled to an area considered endemic for malaria within the time frame for deferral according to your SOP (e.g., less than 1 year prior to donation) and did not report this at the time of donation.
- Donor traveled to an area at risk for vCJD (Variant Creutzfeldt-Jakob Disease) within the time frame for deferral according to your SOP and did not report this at the time of donation.
- When reporting this type of post donation information, you should include in the BPD report all distributed products collected after the implementation of questions used to elicit this information even if you did not conduct notification or retrieval for those products (for example, the report would include Platelets, even if you did not conduct notification or retrieval because the Platelets were expired by the time you received the post donation information).
- Donor or third party provides information of a post donation illness, including, fever (with or without other symptoms), diarrhea, or diagnosis of a specific disease (e.g., mononucleosis, measles, Lyme disease, chicken pox, shingles). Post donation cold or flu symptoms are excluded from reporting.
- Donor later tells the blood establishment that his/her blood should not be used.
- Donor later tells the blood establishment that he/she donated to be tested for a disease or infectious agent (such as HIV).
- Donor provides information of probable or confirmed West Nile Virus infection.
- Donor provides disqualifying information as a result of newly implemented donor history questions and previously collected products were distributed. The donor would have been previously acceptable according to all procedures in place at the time and would not have been deferred even if the donor provided the newly disqualifying information. Examples include:
- Implementation of new deferral criteria related to travel to a vCJD risk area;
- Changes in areas identified by CDC (Center for Disease Control) as malarial endemic.
- Donor provides information of cold or flu symptoms to include any of the following symptoms: general malaise, cough, headache, fatigue, congestion, nausea, vomiting, chills, or runny nose that developed after the donation. The safety, purity, or potency is generally not affected by post donation cold or flu symptoms provided the donor screening process is adequate to detect and defer a donor who presents with any of these symptoms at the time of donation.
- Donor provides information that she was pregnant at the time of donation. This is a donor safety issue and not one that affects the quality of the product.
- Donor Screening and Deferral
- Interview process
- Donor provided information that warranted deferral, but the donor was accepted.
- Donor provided partial information that was not resolved by follow-up questioning, and the donor was accepted.
- Medical evaluation
- Donor's hemoglobin or hematocrit was unacceptable.
- Donor's temperature was unacceptable.
- Plateletpheresis donor had an unacceptable platelet count and there was no documented platelet count for the product.
- Medical review or physical was not performed or was inadequate.
- Donor record documentation
- No documentation of donor's hemoglobin, hematocrit, temperature, arm inspection.
- Answers to the donor history questions, including high risk questions, were missing or incomplete, except those only affecting donor safety (e.g., history of pregnancy or heart disease).
- Donor's signature was missing.
- Donor record was incomplete or incorrect, for example:
- Both confidential unit exclusion stickers were applied to donor record;
- Confidential unit exclusion stickers were applied by person other than donor.
- Deferral procedures
- Donor was incorrectly omitted from the deferral list, and donor subsequently donated.
- Deferral list contained inaccurate information, such as temporary deferral instead of permanent deferral, and the donor subsequently donated.
- The donor tested positive for a viral marker, an appropriate reentry algorithm was not followed and the donor was subsequently allowed to donate.
- The incorrect donor identification information was used to check the deferral list (regardless of whether the donor was previously deferred). You would report even if the donor provided discrepant identification information, such as different names on two donations – J. Michael Smith and James M. Smith.
- The deferral list was not checked (regardless of whether the donor was previously deferred).
- Autologous donor did not meet the suitability criteria and the medical director authorized acceptance of the donor, provided the product was labeled appropriately and not crossed over for allogeneic use.
- Donor did not meet suitability criteria related to donor safety only, such as donor's weight, age, donating within 56 days of last donation, or more than 24 pheresis donations within 12 months. These criteria do not affect product quality.
- Plateletpheresis donor had an unacceptable platelet count, but the platelet count for the product was acceptable.
- A recordkeeping deviation that would not affect the safety, purity, or potency of the product, such as the second or supervisory review of the blood donor record, was not performed or documented, but the initial review was acceptable.
- The deferral list was not checked at the time prescribed in your procedures, or incorrect information was used to check deferral list and you determined prior to distribution of products from that donation that the donor was not previously deferred.
- Donor was accepted on the basis of incomplete information and you obtained additional information from the donor, prior to distribution of any products, demonstrating that the donor was acceptable.
- Donor was deferred and products were handled appropriately, donor was not placed on the deferral list, but the donor did not have subsequent donations, therefore there were no products affected.
- Collection
- Product was contaminated or potentially contaminated with bacteria or air. (The manufacturer that had control of the product during collection would be responsible for reporting this.)
- Arm preparation was not performed or was performed incorrectly.
- Outdated bag or collection set was used in collection.
- Outdated or incorrect anticoagulant was used in collection.
- Defective device or collection bag was used for collection.
- Donor was over bled and the product was distributed as Whole Blood. (Product may be affected if there was an inadequate volume of anticoagulant in the collection bag).
- Source Plasma for further manufacture into injectable products from two different donors was pooled into one pooling bottle.
- *Product was discovered to be clotted.
- **Product was discovered to be hemolyzed.
- Collection specifications were not met, for example:
- Collection time was extended;
- Collection time was discrepant;
- Collection time was not documented;
- Collection status (e.g., satisfactory) was not documented.
- A recordkeeping deviation that would not affect the safety, purity, or potency of the product, such as the phlebotomist's signature was missing from the donor record.
- Donor has a reaction during the collection procedure. If the donor has a fatal reaction as a result of a complication of blood collection, a BPD report is not required, but the collection facility must report to CBER in accordance with the requirements for reporting fatalities (21 CFR 606.170(b)).
- Component Preparation
- Component, such as Platelets or Fresh Frozen Plasma, was not prepared within the appropriate time frame after collection.
- Product was contaminated with bacteria, air or other contaminants during component preparation or processing, such as pooling. (The manufacturer that had control of the product during the component preparation process would be responsible for reporting this.)
- Component was not manufactured according to established procedures, which may affect the safety, purity, or potency for that particular product, such as:
- Platelets were prepared from a product collected from a donor who took aspirin or other drugs that affects platelets;
- Fresh Frozen Plasma or Cryoprecipitated AHF did not meet freezing time requirements;
- Platelets did not meet resting time requirements;
- Component manufactured from a Whole Blood unit that did not meet specifications, such as;
- Overweight or underweight Whole Blood unit,
- Whole Blood unit was collected or stored at unacceptable temperature,
- Extended or undocumented collection time of a Whole Blood unit or collection was difficult or slow;
- Incorrect filter was used for leukoreduction;
- Product was not leukoreduced within the appropriate time frame;
- Incorrect dosage was used for irradiation;
- Product was not washed/deglycerolized properly.
- A recordkeeping deviation that would not affect the safety, purity, or potency of the product, such as the signature of the person preparing the product was missing.
- Testing
- Testing was not performed in accordance with test manufacturer's instructions, such as
- Incorrect incubation time or temperature was used;
- Incorrect reagent was used/licensable components from two different test kit lots were used;
- Incorrect addition of reagents (incorrect sequence, volume or concentration).
- Initial reactive was not repeated in duplicate (viral marker testing), when required by test manufacturer's instructions.
- Sample was tested into compliance (repeated testing until you obtained negative result).
- Test assay was inappropriately invalidated.
- Sample that did not meet specifications for testing was used, such as
- Sample was improperly stored;
- Sample was diluted (e.g., saline dilution of samples collected after an apheresis procedure);
- Sample was not identified appropriately to relate back to donor or patient being tested.
- Testing was performed using a reagent or test kit in which QC was unacceptable or not documented. This may include QC on ABO/Rh typing reagents not documented one day, even if the QC the day before and the day after was acceptable.
- Testing was performed using expired reagents.
- Patient samples were mislabeled or collected from the wrong patient, if you used the sample to crossmatch a product that you distributed. This includes samples collected by transfusion service personnel and personnel outside of the blood establishment.
- Mistyped or misinterpreted patient samples if the sample was used in crossmatching a product that you distributed.
- Immediate spin crossmatch performed when a patient's history or testing requires an indirect antiglobulin test.
- A recordkeeping deviation that would not affect the safety, purity, or potency of the product such as a failure to make one testing record if there are other testing records to indicate that you performed testing appropriately.
- You appropriately invalidated an assay, and you retested samples and they tested negative before product was distributed.
- Labeling
- the product label;
- tie tags;
- the circular of information;
- the labeling accompanying the product that identifies the patient for whom it has been crossmatched (transfusion record).
- Labeling indicated an incorrect ABO or Rh type, antigen type, antibody, anticoagulant, volume or weight.
- Information is missing – ABO or Rh, expiration date, volume, weight, platelet count (for platelet products).
- Labeling indicated testing was performed, but the product was not tested.
- Product was tested but labeling showed testing information that is not consistent with the test results.
- Labeling indicated an incorrectly extended expiration date, even if the product was transfused within the correct dating period.
- Autologous labeling is missing or showed incorrect identifying information, such as the patient's name, social security number, or date of birth.
- Labeling indicated product as crossmatch compatible when the product was either not crossmatched or found incompatible.
- Labeling indicated incorrect recipient name or identification number.
- Labeling indicated an incorrect donor or unit number or donor or unit number is missing. This includes:
- Two units crossmatched for the same patient, but unit identification is switched;
- Unit numbers of split or aliquot products labeled with an incorrect part identifier.
- Labeling indicated an incorrect product name or product name is missing, for example:
- Plasma labeled as Platelets;
- Irradiated Red Blood Cells labeled as Red Blood Cells;
- Red Blood Cells labeled as Red Blood Cells Leukocytes Reduced, but product was not leukoreduced.
- Labeling indicated a shortened expiration date.
- Any of the following information missing from or incorrectly stated on the label (provided the product is otherwise acceptable): collection date (provided the expiration date is correct), or facility identification.
- Unlicensed product labeled with a license number.
- Quality Control and Distribution
- Quality control procedures were not followed or not performed.
- Distribution of product that failed to meet release criteria or was found unacceptable. For example:
- Product was identified as not meeting specifications due to a deviation or unexpected event in donor suitability, collection, component preparation, testing, or labeling and product was distributed;
- Product was distributed prior to determining whether the product was suitable for distribution;
- Events associated with the physical distribution of the product; or
- Additional information reveals product to be unacceptable.
- Product was distributed that should not have been distributed due to:
- Unsuitable donor medical history;
- Incorrect or incomplete testing;
- Positive testing;
- Testing not performed or not documented;
- Outdated product;
- Overweight or underweight unit/component;
- Component prepared from the same Whole Blood unit in which one of the products was determined to be clotted or hemolyzed;
- Licensed establishments: You made a change from what is in your approved biologics license application and you did not comply with the requirements of 21 CFR 601.12 (e.g., distributed products prior to approval of a supplement), if the change may affect the safety, purity or potency of the product. (See, "Guidance for Industry: Changes to an Approved Application: Biological Products: Human Blood and Blood Components Intended for Transfusion or for Further Manufacture," July 2001. (http://www.fda.gov/cber/gdlns/bldchanges.pdf)
- Antibody screen or identification testing was not performed or was incomplete on recipient prior to distribution of product, when required by your SOP;
- Product was not tested for an antigen, which corresponds to the patient's antibody, when required by your SOP.
- Shipping or storage events:
- Product was shipped or stored at incorrect temperature;
- No documentation that product was shipped or stored at appropriate temperature;
- Temperature of a shipment was not recorded upon receipt, and product was redistributed;
- Shipment exceeded time allowed for shipping;
- Product was shipped in an unvalidated container.
- Autologous product distributed as an allogeneic product, but the product did not meet all allogeneic requirements.
- Product was distributed that did not meet all specifications for distribution.
- Product was distributed that was processed using an instrument or reagent in which QC was unacceptable.
- Product was distributed that was processed using an instrument or reagent in which QC was not documented or not performed. Examples of reportable events include:
- No documentation that weekly QC was performed, even if daily QC tasks were performed;
- No documentation of QC trip scales, unless product was weighed prior to distribution and was acceptable.
- Product was distributed in which quality control or product specifications were not met, incompletely documented or not documented. For example:
- pH (monthly QC of platelets);
- Platelet count;
- White blood cell count (for leukoreduced product);
- Red Blood Cell recovery; or
- Bacterial Detection (when positive culture or positive screen using a system approved for QC or pre-release testing of blood products).
- Product distributed prior to resolution of a discrepancy in manufacturing, such as testing, labeling or donor suitability.
- Incorrect product or unit issued for a specific patient (unless the transfusion service notified the hospital staff that the product ordered was unavailable and that the transfusion service was providing a substitute):
- Special processing or testing requested, such as leukoreduced, irradiated, CMV (cytomegalovirus) negative, but product didn't meet specification
- Unit issued that was designated for different patient
- Incorrect product type issued, such as Platelets instead of Fresh Frozen Plasma
- Product with the improper ABO or Rh type was selected for a patient.
- Wrong filter was issued for use in transfusion or a filter was not issued when required.
- Visual check of product was not performed prior to distribution.
- Product was distributed based on testing that was not performed on a current sample.
- Product was accepted as suitable from a blood establishment, and the product was distributed , however it was discovered to be hemolyzed after distribution by the transfusion service.
- Product was shipped to the incorrect facility.
- Product is properly labeled but the shipping invoice differs from the actual shipment.
- Blood establishment shipped an order to another blood establishment for a specific type of product, but did not fulfill the specific request, and the product was properly labeled and not labeled for a specific patient. For example, an order for Red Blood Cells Irradiated was filled with a unit of Red Blood Cells that were properly labeled as Red Blood Cells.
- Donor provides information regarding post donation cold or flu symptoms and products were not quarantined.
- Otherwise unsuitable product, such as an expired HLA (human leukocyte antigen) matched platelet, was issued through emergency release procedures and documented appropriately.
- Frozen product bag (e.g., Fresh Frozen Plasma) broke during thawing and the product was discarded.
- *Product broke or was damaged during shipment and the product was discarded.
- Unlicensed product was distributed labeled with a license number.
- Licensed establishments: You made a change from what is in your approved biologics license application, such as a minor labeling change, and you did not comply with the requirements of 21 CFR 601.12(f), provided the change does not affect the safety, purity or potency of the product (e.g., change in the legal name of the applicant not reported in accordance with 21 CFR 601.12(f)) .
- A segment was found to be clotted or hemolyzed but the product was subsequently evaluated for clots or hemolysis and found acceptable.
- Small residual clots are found in the filter after product was completely transfused, unless you did not adequately evaluate the unit for clots prior to issuing unit for transfusion.
- No documentation of instrument or reagent QC if there was data from another source that showed the instrument or reagent was acceptable, for example:
- No documentation of acceptable reagent storage, but reagent QC was documented to be acceptable;
- Temperature was not documented on recording chart, but manual temperature readings were documented to be acceptable.
- Inappropriate administration practices by the hospital staff in transfusing the patient;
- Hospital staff transfused the wrong patient;
- Hospital staff transfused the patient with the wrong product or unit;
- Hospital staff transfused the patient without using the appropriate filter.
- Allogeneic product was distributed when an autologous product was available.
- Product was distributed using emergency protocol, provided it was labeled appropriately.
- Miscellaneous
- Donor was identified as source of transfusion-associated disease, such as, hepatitis, HIV, malaria, babesiosis, West Nile Virus.
- Donor tested negative and products were distributed. The donor returned and tested confirmed positive at your establishment for a viral marker (HIV, HBV or HCV) for which we require or recommend lookback (21 CFR 610.46 for HIV Lookback, and "Recommendations for the Quarantine and Disposition of Units from Prior Collections from Donors with Repeatedly Reactive Screening Tests for Hepatitis B Virus (HBV), Hepatitis C Virus (HCV) and Human T-Lymphotropic Virus Type I (HTLV-I)", July 19, 1996)
- Recipient identified with transfusion-associated disease, but donor ruled out as source of transfusion-associated disease and donor screening, testing, etc., was acceptable.
- REFERENCES
- Biological Products: Reporting of Errors and Accidents in Manufacturing; Proposed Rule (62 FR 49642, September 23, 1997).
- Biological Products: Reporting of Biological Product Deviations in Manufacturing; Final Rule (65 FR 66621, November 7, 2000).
- Memorandum to All Registered Blood Establishments: "Responsibilities of Blood Establishments Related to Errors & Accidents in the Manufacture of Blood & Blood Components," March 20, 1991.
- Memorandum to All Registered Blood and Plasma Establishments: "Guidance Regarding Post Donation Information Reports," December 10, 1993.
- Guideline for Quality Assurance in Blood Establishments, July 11, 1995.
- Guidance for Industry: Biological Product Deviation Reporting for Licensed Manufacturers of Biological Products Other than Blood and Blood Components, October 2006.
- Guidance for Industry: Changes to an Approved Application: Biological Products: Human Blood and Blood Components Intended for Transfusion or for Further Manufacture, July 2001.
- Memorandum to All Registered Blood and Plasma Establishments: "Recommendations for the Quarantine and Disposition of Units from Prior Collections from Donors with Repeatedly Reactive Screening Tests for Hepatitis B Virus (HBV), Hepatitis C Virus (HCV) and Human T-Lymphotropic Virus Type I (HTLV-I)," July 19, 1996.
This guidance document provides you, a blood or plasma establishment, with the FDA's current thinking related to the biological product deviation (BPD) reporting requirements. For the purposes of this document, "blood and plasma establishment" includes licensed manufacturers of blood and blood components, including Source Plasma, unlicensed registered blood establishments, and transfusion services. This guidance document finalizes the draft guidance of the same title dated August 2001.
FDA's guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidances describe the Agency's current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidances means that something is suggested or recommended, but not required.
Previously, FDA regulations required licensed blood and plasma establishments to report promptly to FDA errors and accidents in manufacturing that may affect the safety, purity, or potency of a product (Title 21 Code of Federal Regulations (21 CFR) 600.14(a) (1999)). On March 20, 1991, we, FDA, issued a memorandum entitled, " Responsibilities of Blood Establishments Related to Errors & Accidents in the Manufacture of Blood & Blood Components" requesting voluntary reporting from unlicensed registered blood establishments and transfusion services.
In the Federal Register of November 7, 2000 (65 FR 66621), we issued a final rule amending the CGMP (current good manufacturing practice) regulations for blood and blood components. The new rule required all establishments involved in the manufacture of blood and blood components, including licensed manufacturers, unlicensed registered establishments and transfusion services, to report product deviations in manufacturing (21 CFR 606.171).1
The new regulation at 21 CFR 606.171, effective May 7, 2001:
Under 21 CFR 606.171, you are required to report certain events associated with the manufacturing, to include testing, processing, packing, labeling, or storage, or with the holding or distribution of blood or a blood component, which may affect the safety, purity, or potency of a distributed product. Safety, purity, and potency are defined in 21 CFR 600.3(p), (r), and (s).
Under 21 CFR 606.171(c), you should submit reports as soon as possible, but you are required to submit reports at a date not to exceed 45 calendar days from the date of discovery of information reasonably suggesting a reportable event has occurred. To facilitate reporting, we have developed a standardized reporting format that you may submit electronically or in paper form, by mail.
The new regulation at 21 CFR 606.171 does not change any of the requirements in 21 CFR Part 606, or Part 211 for conducting investigations of an unexplained discrepancy or the failure of a lot or unit to meet any of its specifications. Those regulations require you to evaluate and investigate, as appropriate, unexplained discrepancies and failures to meet specifications, and to maintain complaint records, including records of investigations and follow-up (21 CFR 606.100, 211.192 and 211.198). We recommend that your procedures for the investigation of any unexplained discrepancy or the failure of a lot or unit to meet any of its specifications include provisions for:
For additional information on investigations of any unexplained discrepancy or failure of a lot or unit to meet any of its specifications, you may refer to the "Guideline for Quality Assurance in Blood Establishments," July 11, 1995, which may be found at http://www.fda.gov/cber/guidelines.htm.
Under 21 CFR 606.171, the manufacturer that had control over the product when the deviation from current good manufacturing practice, applicable regulations, applicable standards, or established specifications or an unexpected or unforeseeable event that may affect the safety, purity, or potency (such a deviation or unexpected, unforeseen event is referred to hereafter as an, "event") occurred must submit a report. This reporting requirement applies to:
We define "control" in 21 CFR 606.3(l ) as having responsibility for maintaining the continued safety, purity and potency of the product and for compliance with applicable product and establishment standards, and for compliance with current good manufacturing practice (CGMP).
If you are a plasma fractionator or IVD manufacturer that collects Source Plasma or other blood components to be used as your source material for further manufacture into a finished product, you are subject to reporting as specified in 21 CFR 600.14(a)(2)(iii). You are required to report, under 21 CFR 606.171(a), events that occur during the manufacture of such source material. If you manufacture that source material into a finished product, you are required to report, under 21 CFR 600.14(a)(2)(iii), events that occur during the manufacture of the licensed finished product (e.g., Immune Globulin Intravenous (Human), Reagent Red Blood Cells).
Occasionally, a blood establishment establishes a contract with another entity to perform some or all of the manufacture of a product. Some common manufacturing steps performed under contract include testing (e.g., viral marker or compatibility), irradiation, blood collection, storage and distribution. If you contract out any manufacturing step, for the purposes of 21 CFR 606.171(a), that step is performed under your control. Under 21 CFR 606.171(a), you must establish, maintain, and follow a procedure for receiving information from that contract manufacturing facility on all deviations, complaints, and adverse events that may affect your products.
For the purpose of 21 CFR 606.171(a), we do not consider contract manufacturing to include the supply of blood products to a transfusion service from a blood establishment or the supply of plasma to a fractionator from a Source Plasma collection establishment.
For the purposes of 21 CFR 606.171(a), we also do not consider a blood establishment to be performing contract manufacturing for an entity that does not perform manufacturing. A hospital would not perform manufacturing if the blood establishment performs the compatibility testing for a hospital, makes the final determination regarding the suitability of a product for a specific patient, and distributes the product to the hospital for transfusion. In this case the blood establishment acts as a transfusion service for the hospital and maintains the patient transfusion record and blood unit disposition history for a hospital. The blood establishment would be responsible for submitting reports related to the transfusion service events.
If you are a contract manufacturer (i.e., under contract, you perform a step in manufacturing for another establishment), you must conduct such manufacturing in accordance with CGMP (21 USC 351(a)(2)(B)), but you are not required to report BPDs to us.
Examples of who must report:
REPORTING
Under 21 CFR 606.100(c), 211.192, and 211.198, the test laboratory must perform an investigation. The test laboratory is NOT required to report to us. The test laboratory would provide the blood establishment with details of the deviation in viral marker testing.
Under 21 CFR 606.171(a), you must establish, maintain, and follow a procedure for receiving information from the test laboratory about deviations concerning the viral testing. Under 21 CFR 606.171(b), you must submit a report to us if you distributed the improperly tested product. We recommend that you assure that the test laboratory performed an adequate investigation.
REPORTING
Under 21 CFR 606.100(c), 211.192, and 211.198, the irradiator must perform an investigation. The irradiator is NOT required to report to us. The irradiator would provide you with details of the deviation in the irradiation process.
Under 21 CFR 606.171(a), you must establish, maintain, and follow a procedure for receiving information from the irradiator about deviations concerning the irradiation process. Under 21 CFR 606.171(b), you must submit a report to us if you distributed the improperly irradiated product. We recommend that you assure that the irradiator performed an appropriate investigation.
REPORTING
Under 21 CFR 606.100(c), 211.192, and 211.198, the irradiator must perform an investigation. Under 606.171(b), the irradiator must submit a report to us if it distributed the improperly irradiated product. You are NOT required to report to us or perform an investigation because the product was not in your control at the time the event occurred, i.e., it was not irradiated under a contract manufacturing agreement.
REPORTING
Under 21 CFR 606.100(c), 211.192, and 211.198, the blood establishment must perform an investigation of the deviation in labeling. Under 21 CFR 606.171(b), the blood establishment must submit a report to us.
You are NOT required to report to us unless contrary to CGMP, applicable regulations, applicable standards, or established specifications, you further distributed the unit without correcting the label.
REPORTING
Under 21 CFR 606.100(c), 211.192, and 211.198, you must perform an investigation of the deviation in labeling. Under 21 CFR 606.171(b), you must submit a report to us if you distributed (i.e., issued for transfusion) the mislabeled product.
REPORTING
Under 21 CFR 606.100(c), 211.192, and 211.198, you must perform an investigation of the inappropriate release of a product with an incorrectly extended expiration date because you failed to recognize the extended expiration date at the time of receipt or compatibility testing. Under 21 CFR 606.171(b), you must submit a report to us because your failure to follow your own SOPs, which would have detected the improper expiration date, was an event that may effect the safety, purity, and potency of the product. We recommend that you notify the blood establishment of the incorrect labeling.
Under 21 CFR 606.100(c), 211.192, and 211.198, the blood establishment must perform an investigation of the deviation in labeling. Under 21 CFR 606.171(b), the blood establishment must also submit a report to us, since the use of an improperly extended expiration date was an event that may affect the safety, purity, and potency of the product.
REPORTING
Under 21 CFR 606.100(c), you must perform an investigation of the improper testing and distribution of the unit. Under 21 CFR 606.171(b), you must submit a report to us. We recommend that you evaluate the need for product retrieval or notification to the fractionator. Note: If your contract-testing laboratory performed the testing, you would be the reporting establishment.
The fractionator would submit a report under 21 CFR 600.14, if it used the improperly tested plasma in the manufacture of a licensed biological product and distributed the final product. The use of the improperly tested plasma would represent an unexpected or unforeseeable event that may affect the safety, purity, or potency of the final product. (See footnote 1)
Under 21 CFR 606.171(b), you must report any event and any information relevant to the event associated with the manufacturing, to include testing, processing, packing, labeling, or storage, or with the holding or distribution, of both licensed and unlicensed blood or blood components, including Source Plasma, if that event meets all the following criteria:
An adequate procedure for deviation reporting (21 CFR 606 . 100(b)) would include steps for determining whether or not an event is one for which a report must be submitted. The decision to report should be based on whether the event had the potential to affect the safety, purity, or potency of a product. The terms safety, purity, and potency are defined in 21 CFR 600.3(p), (r), and (s), respectively.
Retrieval, Consignee Notification and Lookback
You are not required to submit a BPD report simply because you failed to follow your own internal procedures for retrieval, notification or lookback (e.g., you do not have to submit a report if you did not notify consignees within the time frame prescribed in your procedures). The failure to follow retrieval, notification, or lookback procedures does not, by itself, affect the safety, purity, or potency of the product. However, under 21 CFR 606.171(b), you must submit a report if the underlying reason for the retrieval, notification, or lookback meets the reporting criteria found in Section III.B. In that case, the report must describe the event that may have affected the safety, purity, or potency of the distributed product and describe the failure to follow procedures. For example, you distributed a product collected from a donor with a history of high risk behavior, which may affect the safety, purity, or potency of the product. Your SOP requires that you notify consignees of the product within 5 days and you notified the consignee 8 days after discovering the event. You are required to report the improper release of the product. We recommend that you also include in the report that consignee notification was not in accordance with your SOP.
You are not required to submit a report:
While the above examples would not be reportable under 21 CFR 606.171, the events may constitute deviations from the regulations, which we would assess during inspection.
Biological Product Deviation Reporting Flow Chart for Blood and Plasma Establishments
You may use the following flow chart to help you determine if you are required to report an event to us.
Biological Product Deviation Flow Chart for Blood and Plasma Estalbishments
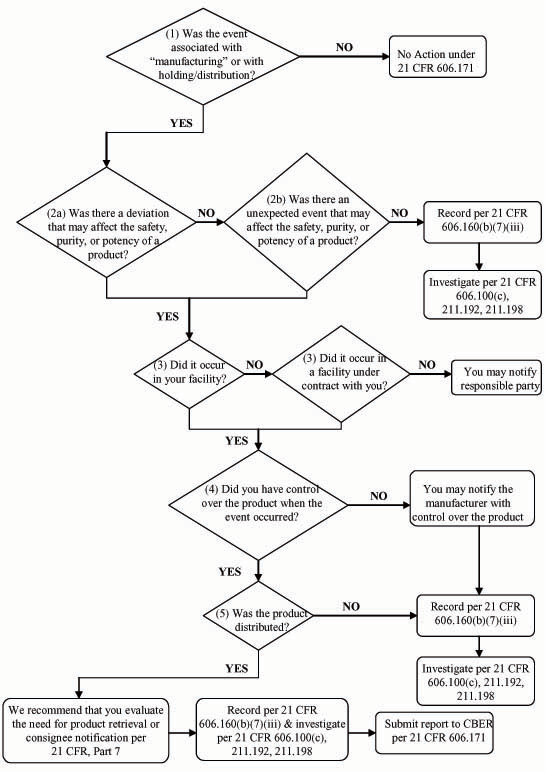
The following questions correspond to the flow chart:
(1) Was the event associated with "manufacturing" or holding or distribution as they are described in the regulation?
As described in 21 CFR 606.171(b), manufacturing includes testing, processing, packing, labeling, or storage. In addition, you must report events associated with the holding or distribution of both licensed and unlicensed blood or blood components, including Source Plasma.
Under 21 CFR 606.171, you are not required to report events that occur after you distribute the product, including those related to the administration of blood or blood components.
Examples
(2a) Was there a deviation that may affect the safety, purity, or potency of a product?
A deviation that may affect the safety, purity, or potency of a product could include any change in the manufacturing process that could prevent a product from meeting all CGMP requirements, applicable standards, and established specifications. The CGMP and applicable regulations for blood and blood components are currently found in 21 CFR Parts 210, 211, 600, 606, 610, 630, and 640. Standards refer to specifications and procedures applicable to the manufacture or release of products, which are established to help ensure the continued safety, purity, and potency of such products. Specifications refer to quality standards (i.e., tests, analytical procedures, and acceptance criteria) that confirm the quality of products and other materials used in the production of a product, e.g., the hematocrit level, or the temperature range for thawing a frozen component.
(2b) Was there an unexpected or unforeseeable event that may affect the safety, purity, or potency of a product?
An unexpected or unforeseeable event is one in which, despite the fact that you followed all required procedures, something occurred that may affect the safety, purity, or potency of a product. This may be due to information that you did not have at the time of manufacturing. Examples of unexpected or unforeseeable events in which the safety, purity, or potency may be affected include the following:
If an event occurred, but could not have affected the safety, purity or potency of a product, 21 CFR 606.160(b)(7)(iii), 606.100(c), 211.192 and 211.198 require that you record and investigate it, but you do not need to report to us.
Under 21 CFR 606.171(b), if you discover an event after you distributed a product and the safety, purity, or potency of the product may have been affected because of the event, you must report the event, regardless of whether consignee notification or product retrieval is necessary. You must report the event even if you ultimately determined, through investigation after distribution, that the safety, purity or potency of the product was not affected.
For example, if you distributed a product that was not tested for all viral markers, under 21 CFR 606.171(b), you must submit a report to us, even if you subsequently tested the product and found it to be negative.
If you discover an event prior to distribution of a product and determined that the event did not affect the safety, purity, or potency of the products, you are not required to report under 21 CFR 606.171.
For example, if you discovered a deviation in testing prior to the distribution of a product and you appropriately retested the product and found it to be negative, you are not required to report to us under 21 CFR 606.171.
(3) Did it occur in your facility or in a facility under contract with you?
You are required to submit a report if the event occurred within your facility or a facility under contract with you, such as a testing laboratory. Under 21 CFR 606.171(a) and (b), you must report events that occur at the contractor and you must establish, maintain, and follow a procedure for receiving information from the contract facility on all deviations, complaints, and adverse events concerning your potentially affected products.
If you are a contract manufacturer, such as a testing laboratory, and an event occurs within your facility, you would notify the manufacturer with control over the product, if its products may be affected. You are not responsible for reporting the event to us.
If you detect an event that occurred in another facility not under contract with you, we recommend that you contact that facility, which would be responsible for reporting to us, if appropriate. For example, if you receive a unit of Red Blood Cells shipped without ice we recommend that you notify the supplier. You are not required to report to us unless, contrary to CGMP, applicable regulations, applicable standards, or established specifications, you further distributed the unacceptable unit.
(4) Did you have control over the product when the event occurred?
You have control over the product if you have overall responsibility for:
You are responsible for reporting if you had control over the product when the event occurred and you distributed the affected product.
You have control over the product if you contract with another entity to perform all or some of the manufacture of a product. Under 21 CFR 606.171(a), you must establish a procedure for receiving information from the contract manufacturing facility on all deviations, complaints and adverse events. The contract manufacturer is responsible for documenting the event in accordance with 21 CFR 606.160(b)(7)(iii) and investigating in accordance with 21 CFR 606.100(c), 211.192 and 211.198. The contract manufacturer is not responsible for reporting to us.
(5) Was the product distributed?
We define "distributed" in 21 CFR 606.3(k) as:
(1) the blood or blood component has left the control of the licensed manufacturer, unlicensed registered blood establishment, or transfusion service; or
(2) the licensed manufacturer has provided Source Plasma or any other blood component for use in the manufacture of a licensed biological product.If you are a licensed blood establishment, a product is considered distributed when you shipped the product to another facility or broker that is not part of your license or issued the product to staff outside the blood establishment for transfusion. A product is not considered distributed if it was shipped from one location to another and both locations are under the same license.
If you are an unlicensed blood establishment, including a transfusion service, a product is considered distributed when you issued the product to staff outside of the blood establishment for transfusion or you shipped it to another facility or broker.
If you are a Source Plasma establishment, the product is considered distributed when you released or shipped the product for further processing. Distribution includes shipment to a plasma broker. Distribution does not include shipment to an off-site storage facility if the product is still under your control.
Examples:
If you distributed the product, we recommend that you also assess the need for product retrieval or consignee notification in accordance with 21 CFR Part 7. You must document the event in accordance with 21 CFR 606.160(b)(7)(iii) and investigate in accordance with 21 CFR 606.100(c), 211.192 and 211.198, regardless of whether you distributed the product.
Under 21 CFR 606.171(c), you should report a biological product deviation as soon as possible, but you must report at a date not to exceed 45 calendar days from the date that you, your agent, or another person who performs a manufacturing, holding, or distribution step under your control, acquire information reasonably suggesting that a reportable event has occurred. You acquire such information when any employee of your facility, not just those involved in quality assurance or quality control activities, learns about the event. As soon as you acquire information, you should make an assessment of whether the event had the potential to affect the safety, purity, and potency of products and determine the status of the products (i.e., whether they have been distributed or need to be quarantined).
If you contract with a facility to perform a manufacturing step and an event occurs at the contractor, the time period for reporting starts when your contractor learns about the event.
Under 21 CFR 606.171(d), you must use Form FDA-3486 to report BPDs. You must submit the completed report either electronically through CBER's web site at http://www.fda.gov/cber/biodev/biodev.htm or by mail to:
Director, Office of Compliance and Biologics Quality (HFM-600)
Center for Biologics Evaluation and Research
1401 Rockville Pike, Suite 200N
Rockville, Maryland 20852-1448
If the event occurred at your contract manufacturer, we recommend that you include in the report the details reported to you by the contract manufacturer regarding the event.
Complete a separate report for each event. If an event involves more than one product, you only need to complete one report listing all distributed products affected.
The Form FDA-3486 and instructions for completing both formats are located at http://www.fda.gov/cber/biodev/biodev.htm
We categorize BPD reports according to the manufacturing system where the event occurred. Following the bulleted list below, each system is explained further and we provide examples of both reportable and non-reportable events. An event may be the result of a failure within a variety of systems. It is important for you to know both where the event occurred and why your product was allowed to continue through the manufacturing process and distributed, so that you can implement the appropriate corrective action to prevent recurrence.
The following examples of events are not all-inclusive and do not represent all variations that may occur. The examples include deviations from CGMP, applicable regulations, applicable standards, and established specifications, in addition to unexpected or unforeseeable events. Not all of these examples will necessarily apply to you or to all products you manufacture. Whether or not the examples apply to you depends on your manufacturing operations and procedures. Under 21 CFR 606.100(c), 211.192, and 211.198, all events must be investigated.
Over the past several years, donor suitability issues have represented the largest percentage of reports. This category includes issues related to post donation information, donor screening, and donor deferral.
The majority of reports related to donor suitability involved post donation information. We issued a Memorandum to All Blood Establishments entitled, "Guidance Regarding Post Donation Information Reports," on December 10, 1993, which described the process for handling post donation information. This guidance does not supersede the memorandum, but provides additional information regarding the reporting of post donation information.
Post donation information includes information that a donor or other reliable source provides to you subsequent to a donation. Post donation information is most often reported at a subsequent donation. Report post donation information if you would have deferred the donor had you known the information at the time of donation and the safety, purity, or potency of the product could be affected. It is also reportable if the medical evaluation reasonably suggests that the safety, purity, or potency of the product could be affected, such as when a donor reports a post donation illness, or the information is insufficient to conclude that the safety, purity, or potency of the product is not affected, such as when a donor subsequently instructs an establishment not to use the donor's blood without providing a reason.
In some cases blood establishments cannot control post donation information, but they are responsible for the safety, purity, and potency of the product and for compliance with applicable standards and CGMP requirements. For example, a donor may call after donating to report a post donation illness, or information obtained post donation about exposure to a disease or a sex partner at high risk. In other cases, the donor has knowledge of a behavior or risk factor prior to donation that would cause the donor to be unsuitable, but the donor fails to report this at the time of donation. At some point after donation, the donor provides information that results in deferral.
Submit a report when you receive post donation information that may affect the safety, purity or potency of a product you distributed. Examples of reportable events associated with post donation information may include:
DO NOT REPORT:
Donor screening and deferral events include those relating to determining donor eligibility, which you did not discover until after you distributed the product. This category includes events concerning the interview or medical evaluation process, blood donor record documentation, and deferral procedures and records.
Under 21 CFR 606.171(b), you must submit a report when there is an event (a deviation or unexpected or unforeseeable event) during the interview process that may affect the safety, purity, or potency of a product you distributed. Examples of reportable events associated with the interview process may include:
Under 21 CFR 606.171(b), you must submit a report when there is an event (a deviation or unexpected or unforeseeable event) during the medical evaluation that may affect the safety, purity, or potency of a product you distributed. Examples of reportable events associated with medical evaluation process may include:
Under 21 CFR 606.171(b), you must submit a report when there is an event (a deviation or unexpected or unforeseeable event) during donor record documentation that may affect the safety, purity, or potency of a product you distributed. Examples of reportable events associated with donor record documentation may include:
Under 21 CFR 606.171(b), you must submit a report when there is an event (a deviation or unexpected or unforeseeable event) during the deferral process that may affect the safety, purity, or potency of a product you distributed. Examples of reportable events associated with the deferral process may include:
DO NOT REPORT:
Collection events include those that occur during the collection process, which you did not discover until after you distributed the product.
Under 21 CFR 606.171(b), you must submit a report when there is an event (a deviation or unexpected or unforeseeable event) during the collection process that may affect the safety, purity, or potency of a product you distributed. Examples of reportable events associated with the collection process may include:
*The manufacturer who had control of the product during collection should report clotted products that were distributed.
**The manufacturer who had control of the product during collection, component preparation and storage should report hemolyzed products that were distributed and were not accepted into the consignee's inventory. A transfusion service would be responsible for reporting hemolyzed products if the product was inspected upon receipt from the collection facility and found acceptable (no hemolysis) and then later found to be hemolyzed while in the control of the transfusion service and subsequently distributed (reported under QC & Distribution system).
DO NOT REPORT:
Component preparation events include those that occur during the preparation or processing of a product, which you did not discover until after you have distributed a product.
Under 21 CFR 606.171(b), you must submit a report when there is an event (a deviation or unexpected or unforeseeable event) during the component preparation process that may affect the safety, purity, or potency of a product you distributed. Examples of reportable events associated with the component preparation process may include:
DO NOT REPORT:
Testing events include those that occur during the testing process, which you did not discover until after you have distributed the product. Testing includes all tests used to assure the safety, purity, and potency of a product, whether required by specific regulations or by your SOP. Product release testing may include testing performed under IND (investigational new drug application). Use of an unsuitable or inappropriate sample may also be a testing deviation or unexpected event.
Under 21 CFR 606.171(b), you must submit a report when there is an event (a deviation or unexpected or unforeseeable event) during testing that may affect the safety, purity, or potency of a product you distributed. Examples of reportable events associated with testing may include:
For transfusion services, reportable events may also include:
DO NOT REPORT:
Labeling events include those that occur during the labeling process, which you did not discover until after you have distributed the product. Labeling events include incorrect, missing or misleading information on any labeling pertaining to the product, including:
Under 21 CFR 606.171(b), you must submit a report when there is an event (a deviation or unexpected or unforeseeable event) during labeling that may affect the safety, purity, or potency of a product you distributed. Examples of reportable events associated with labeling may include:
DO NOT REPORT:
Events in quality control and distribution are those in which:
Under 21 CFR 606.171(b), you must submit a report when there is an event (a deviation or unexpected or unforeseeable event) in quality control/quality assurance or in the distribution process that may affect the safety, purity, or potency of a product you distributed. Examples of reportable events associated with quality control/assurance or distribution process may include:
For transfusion services, reportable events may also include the distribution of a product in which:
For transfusion services, reportable events may also include the distribution of products in which specifications were not met, which may affect the safety, purity, or potency of a product, such as:
DO NOT REPORT:
* Broken or damaged products are rarely identified as system problems in manufacturing and are generally tied to an unusual event in shipment.
For transfusion services, non-reportable events may also include:
Other situations that would be reportable as an unexpected or unforeseeable event that may affect the safety, purity, or potency of previously distributed products include:
NOTE: You may not need to include all previous donations in the report. The report would include only those products subject to lookback, as identified in the regulation or recommended in guidance.
DO NOT REPORT
1 Under 21 CFR 600.14, manufacturers of licensed biological products other than blood and blood components, including vaccines, allergenic products, therapeutics, plasma derivatives, and in vitro diagnostics (IVD), are required to submit reports in a manner similar to the requirements in 21 CFR 606.171. We have issued a separate guidance for reporting of product deviations by licensed manufacturers of biological products other than blood and blood components (Guidance for Industry: Biological Product Deviation Reporting for Licensed Manufacturers of Biological Products Other than Blood and Blood Components, October 2006). This guidance document is available through the Internet at http://www.fda.gov/cber/guidelines.htm or through CBER's Office of Communication, Training, and Manufacturers Assistance.