Computing Life

Overview of NIGMS and Computing Life
Searching for Genetic Treasures
The Next Top Protein Model
Movie Mania
Sim Sickness
Integrating Biology
Made Possible By ...
Web Extras: Movies, Podcasts, and Other Cool Stuff
"Computing Life" in PDF
Web Extras: Article
Now Open for Drug Chemistry
The antibiotic penicillin was discovered by accident when mold happened to drift in from another lab, land on Alexander Fleming’s petri dish, and kill bacteria on it. But not all drug discoveries happen so easily. Researchers can spend years searching for the right compound before it even reaches a clinical trial.
With improvements in information technology, pharmaceutical scientists don’t need to wait for something to fall into their petri dish. Computational tools can help them quickly identify the compounds with the most therapeutic promise, making drug discovery faster and cheaper.
But as with most technologies, there’s always room for improvement. By getting back to the basics, a new project plans to give computer-aided drug design a boost.
Heather Carlson, a chemist in the College of Pharmacy at the University of Michigan, will spend the next five years gathering the best possible data and making it freely available via the Internet to anyone who wants to advance the computational tools needed for drug discovery.
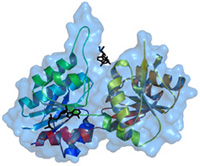
Finding a compound that could be a drug is like solving a puzzle—one with tens of thousands of possible pieces. Carlson says that just as you’d try to fit a puzzle piece based on its shape and color, pharmaceutical scientists look for compounds with a particular shape and other features that “fit with” a target protein.
Fortunately, computers play the part of puzzle solver. Software programs let researchers predict which among all those drug-like molecules not only bind to a target protein, but do so in a potentially useful way. These may be candidates for drug development.
But to get the right answer, Carlson says, you need the best data, which currently is very limited. “Computational techniques do a good job—they’ll tell you if a molecule is in the ballpark,” explains Carlson. “But to find a molecule that leads to a new drug, they’ve got to be more exact.”
So her goal for the new resource is to make the finest data available for others to build computational tools that more precisely identify drug candidates. The resource will include molecular details about proteins attached to small, drug-like molecules called ligands. Ligands can latch onto a protein and alter a biological process. Drugs work in much the same way.
Carlson already has a strong start on the project. She will draw from her own database called the Binding MOAD (“Mother of All Databases”), which contains the structures of more than 11,000 protein-ligand complexes. And, she’s collaborating with another scientist who developed a resource of binding data.
The challenge, she says, will be getting new data. With 10 people working beside her, Carlson will collaborate with chemists and structural biologists to generate some data themselves. They’ll also track down unpublished results, hoping to get many from pharmaceutical company scientists who until now haven’t disclosed their findings. By sponsoring meetings and workshops, they hope to spark more interest in sharing and producing new data.
“We’re really going to depend on community participation,” says Carlson, who wants the resource to foster an inclusive, collaborative research environment. “I really hope our effort gets embraced by everyone working in this field.”
If it does, computational chemists may be closer to realizing their dream: Plugging a disease-related protein into a computer and getting out a new drug.