Research
Artur Ramos,*† Amilcar Tanuri,† Mauro Schechter,† Mark A. Rayfield,*
Dale J. Hu,* Maulori C. Cabral,† Claudiu I. Bandea,* James Baggs,‡ and Danuta
Pieniazek*
*Centers for Disease Control and Prevention, Atlanta, Georgia, USA; †Universidade
Federal do Rio de Janeiro, Brazil; ‡Emory University, Atlanta, Georgia, USA
| We systematically evaluated multiple and recombinant infections in an HIV-infected population selected for vaccine trials. Seventy-nine HIV-1 infected persons in a clinical cohort study in Rio de Janeiro, Brazil, were evaluated for 1 year. A combination of molecular screening assays and DNA sequencing showed 3 dual infections (3.8%), 6 recombinant infections (7.6%), and 70 (88.6%) infections involving single viral subtypes. In the three dual infections, we identified HIV-1 subtypes F and B, F and D, and B and D; in contrast, the single and recombinant infections involved only HIV-1 subtypes B and F. The recombinants had five distinct B/F mosaic patterns: Bgag-p17/Bgag -p24/Fpol/Benv , Fgag-p17/Bgag -p24/Fpol/Fenv , Bgag-p17/B-Fgag -p24/Fpol/Fenv , Bgag-p17/B-Fgag -p24/Fpol/Benv , and Fgag-p17/B-Fgag -p24/Fpol/Fenv . No association was found between dual or recombinant infections and demographic or clinical variables. These findings indicate that dual and recombinant infections are emerging as an integral part of the HIV/AIDS epidemic in Brazil and emphasize the heterogenous character of epidemics emerging in countries where multiple viral subtypes coexist. |
Our understanding of the global molecular epidemiology of HIV-1 infections has improved substantially in recent years. Remarkable antigenic diversity, especially in the viral envelope, has emerged among HIV isolates worldwide (1). As the HIV/AIDS pandemic grows, viral strains are becoming more geographically dispersed, and the simultaneous presence of multiple subtypes in a given region is now common (2). Mixed infections and recombinants involving sequences of distinct HIV-1 subtypes (mosaics) are being recognized, but their prevalence and effect on the pandemic have not been fully evaluated (3). Consequently, the distribution of dual infections and mosaic viruses among different populations and the changes of this distribution over time are still relatively unknown. In addition, scientists are increasingly interested in possible differences in the transmission, epidemiologic patterns, and natural history of HIV-1 infections caused by more than one viral subtype and recombinant genomes. Finally, the efficacy of HIV-1 vaccines, primarily developed against subtype B viruses, may differ against more divergent recombinant variants and mixed infections of distinct HIV-1 subtypes. Such knowledge is vital to understanding the relevant role of mixed infections as a prerequisite for recombination and could be applied immediately in molecular epidemiology and immunotherapy.
Studies using convenience samples first documented mixed infections caused by viruses of subtypes B and E in Thailand (4). Subsequently, such studies have identified cases of dual infections with subtypes B, F, C, and D in Brazil (5-7), B and F in Puerto Rico (8), A and C in Rwanda (9), and triple HIV-1 infection with groups O and M of different clades in a single Cameroonian AIDS patient (10). In addition, potential dual infections have been detected by molecular screening assays among HIV-1 infected populations in Uganda and Kenya, where subtypes A and D coexist (11). The consequences of HIV-1 mixed infections may profoundly influence the dynamic of the pandemic through altered patterns of viral transmission and pathogenesis. Moreover, the resulting genetic variation may lead to the emergence of new HIV variants, including those with altered antigenicity and reduced sensitivity to detection by current diagnostic assays. The global emergence of such variants is exemplified in the impact of two HIV-1 recombinants of subtypes A/E and G/A on epidemics in Thailand and in certain parts of Central Africa, respectively (1,12-14). Interestingly, the presumptive parental subtypes E and G have not been identified.
Because Brazil has been selected as a World Health Organization field site for HIV-1 vaccine evaluation programs, priority has been given to extensive molecular examination of the prevalence and genetic diversity of HIV-1 strains circulating in the country. By November 1997, 116,277 AIDS cases had been reported to the Brazilian AIDS Control Program of the Ministry of Health (15). The 293 HIV-1 strains that have been molecularly characterized document that, although four HIV-1 subtypes B, F, C, and D are circulating in Brazil, only subtypes B and F are common (16-21). Moreover, five HIV-1 dual infections were identified in 21 HIV-infected patients during testing of molecular techniques that would discriminate between HIV-1 infections caused by single and multiple subtypes (5-7). Also, two cases of B/F recombinants have been found accidentally through sequence analysis of the env region (22). These data only indicate the potential for dual and recombinant infections in Brazil, where multiple subtypes circulate; they cannot, however, be used to assess the frequency of these infections among the HIV-infected population. In this study, we evaluated the proportion of HIV-1 dual and recombinant infections among 79 patients enrolled in a prospective clinical cohort study in Rio de Janeiro (an area where HIV-1 subtypes B and F are common).
Population
Part of an ongoing prospective clinical cohort study established in 1991 by the AIDS program of the Federal University of Rio de Janeiro, the HIV-1-infected patients have been continuously enrolled in the cohort for consultation, treatment, evaluation of different clinical parameters during the progression of the disease, and assessment of the genetic variation of HIV-1 strains (23). With informed consent, blood samples were obtained from the 79 patients, who consecutively attended the clinic between October and December 1994. For the purposes of this study, samples collected in 1994 were compared as needed, and follow-up specimens were collected 1 year later. The clinical profile of patients was based on the medical evaluation at the time of blood collection. Because the patients were randomly selected, the findings presented in this article likely reflect the trends in the HIV/AIDS epidemic in the Rio de Janeiro area. Epi Info, Version 6 program (CDC, Atlanta, GA, USA) was used to calculate frequencies, means, and analyses of variance of demographic information, clinical stages, and laboratory parameters.
Design
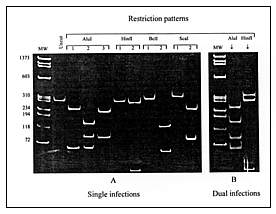 |
Click here to view enlarged imageFigure 1. Differentiation between single (A) and dual (B) HIV-1 infections by the restriction fragment length polymorphism analysis of the polymerase chain reaction—amplified prt. A: Three AluI digestion patterns represent subtypes A, C, and F (pattern 1) and subtypes B and D (patterns 2 and 3); two HinfI patterns represent subtypes D (pattern 1) and B (pattern 2); two BclI patterns represent subtypes F (patterns 1) and A and C (pattern 2); two ScaI patterns represent subtypes A (pattern 1) and C (pattern 2). B: Two AluI digestion patterns (1 and 2) in the dually infected patient with HIV-1 subtypes F and B; two HinfI patterns (1 and 2) in the patient infected with subtypes B and D viruses. |
To investigate the proportion of mixed infections, we must distinguish between those involving two or more distinct HIV-1 strains and infection with a single intersubtype recombinant strain. By definition, the mixed infection occurs when multiple phylogenetically distinct copies of the same gene representing different viral genomes are present within one patient (24). In contrast, in the mosaic strain, different viral regions within the same genome are classified by phylogenetic analysis into different subtypes (3). Potential HIV-1 mixed infections involving distinct HIV-1 variants were segregated from single infections caused by only one subtype by using a restriction fragment length polymorphism (RFLP) screening assay of the prt gene (5,7). The restriction map profiles of the viral prt allow the segregation of HIV-1 strains to subtypes A, B, C, D, and F (Figure 1A). AluI digestion patterns separate subtypes A, C, and F from subtypes B and D. Sequential restriction analysis of prt using HinfI, BclI, and ScaI restriction enzymes further differentiates among these two subtype groups. The simultaneous occurrence of more than one digestion pattern indicates a potential mixed infection (Figure 1B). We selected recombinants among RFLP-subtyped single infections by additionally subtyping the C2-V3 env region with the heteroduplex mobility assay (25). Subtype discrepancies between prt and env regions were considered potential HIV-1 recombinants. All potential multiple infections and recombinants were analyzed by sequence analysis. The search for recombinants was further expanded by sequence analysis of the entire p17 gag or a 311-bp of the p24 gag fragment or both among preselected prt-env recombinants, all prt-env subtype F, and some prt-env subtype B viruses.
Polymerase Chain Reaction (PCR)
Uncultured or cultured peripheral blood mononuclear cells from patients were used for nested-PCR of the entire HIV-1 protease gene (prt, 297-bp), p24 gag fragment (311-bp), and C2-V3 domain of env (565-bp) (6,7). The outer primers for amplification of a 717-bp gag fragment spanning the entire 17 gag region and a 311-bp of p24 gag were LTRF: 5'GGGCTAATTTGGTCAAAAAGAAG; nucleotide position: 6-28, HIV-1MN, and P24RA: 5'ATGTCACTTCCCCTTGGTTCT; nucleotide position: 1482-1502. The inner primers were P17F: 5'GCAAGAGGCGAGGGGCAGCAGCCG; nucleotide position: 716-739, and P17R: 5'CCCATTCTGCAGCTTCATTGA; nucleotide position: 1413-1433. The outer primers for amplification of a 1,444-bp fragment from p24 gag to the reverse transcriptase region were P24F1: 5'ATAGAGGAAGAGCAAAACAAAA; nucleotide position: 1,099 to 1,120, and MOPR1: 5'AAAATTGGAGTATTGTATGGATT; nucleotide position: 2,724 to 2,746. The inner primers were P24FA: 5'CAAAATTACCCTATAGTGCA; nucleotide position: 1,177 to 1,196, and MOPR2: 5'GGTCCATCCATTCCTGGTTT; nucleotide position: 2,601 to 2,620. PCR conditions were the same for amplification of all viral regions (7).
Sensitivity of Detection of Dual Infections
| Table 1. Sensitivity of detection of HIV-1 dual infections caused by viruses of subtypes B and F | ||||||
| No. of viral subtype |
Ratio F:B |
No. of experi- ments |
AluI digestion patterns of prt |
|||
| F | B | B | F | B&F | ||
| 100 | 100 | 1:1 | 1 | – | – | + |
| 50 | 50 | 1:1 | 4 | – | – | + |
| 25 | 25 | 1:1 | 4 | – | + (1) | + (3) |
| 10 | 10 | 1:1 | 4 | – | – | + |
| 5 | 5 | 1:1 | 4 | – | + (3) | + (1) |
| 100 | 5 | 20:1 | 2 | – | + (1) | + (1) |
| 100 | 10 | 10:1 | 2 | – | + (1) | + (1) |
| 100 | 25 | 4:1 | 2 | – | + | – |
| 100 | 50 | 2:1 | 2 | – | – | + |
| 5 | 100 | 1:20 | 1 | – | – | + |
| 10 | 100 | 1:10 | 1 | – | – | + |
| 25 | 100 | 1:4 | 1 | – | – | + |
| 50 | 100 | 1:2 | 1 | – | – | + |
| 100 | 0 | 3 | – | + | – | |
| 50 | 0 | 3 | – | + | – | |
| 25 | 0 | 3 | – | + | – | |
| 10 | 0 | 3 | – | + | – | |
| 5 | 0 | 2 | – | + | – | |
| 0 | 100 | 3 | + | – | – | |
| 0 | 50 | 2 | + | – | – | |
| 0 | 25 | 3 | + | – | – | |
| 0 | 10 | 3 | + | – | – | |
| 0 | 5 | 3 | + | – | – | |
| Absence (-) and presence (+) of AluI digestion pattern of prt characteristic for subtype B, subtype F, and combination of subtypes B and F (Fig. 1). The cloned proviral DNA of HIV-1 subtypes B and F spanning a 1444-bp fragment from p24 gag to rt was used for the nested PCR amplification of prt. Amplified products were digested with AluI restriction enzyme, and the presence of two digestion patterns was analyzed on a 10% polyacrylamide gel by ethidium-bromide staining. | ||||||
To determine the sensitivity of detection of dual infections by the RFLP assay, we mixed 5, 10, 25, 50, and 100 copies of subtypes B and F cloned proviral DNA in equal proportions or in ratios 1:2, 1:4, 1:10, and 1:20 for nested PCR amplification of prt (Table 1). For each combination, one to four independent PCR reactions were run. The simultaneous amplification of the viral prt of subtypes B and F was evaluated by the RFLP assay. PCR controls included DNA templates of single HIV-1 subtypes.
Cloning, Sequencing, and Phylogenetic Analysis
The PCR-amplified proviral prt sequences from potential dual infections were cloned by using the Original TA Cloning Kit (Invitrogen, Carlsbad, CA, USA). DNA from 30 clones of each specimen was screened for distinct HIV-1 sequences by the RFLP assay (7). Double-stranded viral DNA from selected clones or from direct PCR-amplified prt, p17 and p24 gag, and C2-V3 env products was cycle-sequenced in both directions with fluorescent dye labeled sequencing terminators (26). Sequencing reactions were run in an automated DNA sequencer (Applied Biosystems, Foster City, CA, USA). The sequences were aligned by the CLUSTAL multiple sequence alignment program (27). After gaps were eliminated, the aligned sequences were analyzed by the maximum likelihood method, with the fastDNAml program, which uses randomized data input and global rearrangement (28). Additionally, the neighbor joining method (PHYLIP package version 3.5c [29]) was used, with or without bootstrapping. The stability of the tree's topology was tested by pruning (removing one sequence from the alignment and rerunning the phylogenetic analysis). The SIV-cpz sequences (GenBank accession no. X52154) were used as outgroups. HIV-1 sequences generated in this study have been submitted to GenBank.
Detection of Mixed Infections
RFLP analysis of the prt gene showed the simultaneous presence of two different digestion patterns in specimens from 10 of 79 patients. A complex pattern composed of elements of AluI patterns #1 (subtypes A, C, or F) and #2 (subtypes B or D) was identified in nine patients, and a combination of two HinfI digestion patterns (subtypes B and D) was found in one patient (Figure 1B). These data suggest that each of 10 patients could be infected with multiple distinct HIV-1 subtypes. The remaining 69 samples were classified as single infections caused by viruses of prt subtypes B (59) and F (10).
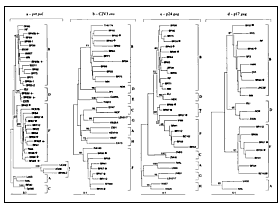
The PCR-amplified viral prt products from these 10 patients were cloned and sequenced to confirm the presence of mixed infections. Sequence analysis showed the simultaneous presence of two distinct HIV-1 variants in only three patients: BR45, BR62, and BR83. The nucleotide divergence between the two prt sequences within the patients was 6.8% for BR45, 7.4% for BR62, and 7.1% for BR83, indicating two distinct HIV-1 variants in each of these patients (6). Phylogenetic analysis confirmed these findings and demonstrated that the divergent HIV-1 prt sequences segregated into subtypes B and F (BR45), subtypes D and F (BR62), and subtypes B and D (BR83) (Figure 2a). In the remaining seven specimens, the observed RFLP results were consequences of either point mutations in AluI restriction site (five cases) or G A hypermutation (two cases), which occurred across one of the sequences within each specimen and destroyed the defining AluI sites. These changes in AluI restriction sites gave rise to genetically distinct quasispecies within the patients but did not represent distinct subtypes, as further confirmed by phylogenetic analysis (e.g., BR55-1 and BR55-2, and BR99-1 and BR99-2 in Figure 2a). Thus, despite the presence of mixed AluI digestion patterns in these seven specimens, they were classified as single infections of subtype B variants.
 |
Click here to view enlarged image
|
To address the issue of potential laboratory contamination, we collected repeat blood samples from dually infected patients approximately 1 year later and processed them on separate occasions. (Blood was unlikely to be contaminated during collection because a disposable vacutainer system was used to obtain each blood sample.) The sequence data from the first and second blood samples of each person showed a 98% to 99% similarity. Also, the viral sequences from these patients were distinct from those of laboratory strains (Figure 2a; 855M, 8,986, and 9,001) commonly used as standards.
Sensitivity of Detection of Dual Infections
To investigate the sensitivity of the RFLP screening method, we performed reconstruction experiments, in which two distinct viral DNA templates of subtypes B and F were analyzed in the same reaction mixture (Table 1). When equal proportions of 10 to 100 HIV-1 DNA template copies were used for prt amplification, two viral subtypes could be simultaneously detected in all but one of 19 experiments. However, when five or fewer copies of each viral subtype were used, two subtypes were identified in only one of four assays; in the remaining three assays either subtype B or subtype F amplicons, but not both, were found. Similarly, in experiments containing varying proportions of subtypes B and F DNA templates, the simultaneous presence of two viral subtypes could be detected in only 8 (66%) of 12 experiments. In comparison, 5 to 100 copies of a single viral subtype (B or F) were routinely amplified and identified in all control reactions.
Detection of Intersubtype HIV-1 Recombinants
Parallel RFLP/heteroduplex mobility assay screening for HIV-1 subtypes in the prt and env regions identified two potential recombinants among 76 single infections. Subtype F prt and subtype B env were found in both specimens BR43 and BR60 during this initial screening. The remaining specimens were classified into subtypes F (n = 8) and B (n = 66) in both prt and env regions. These prt-env potential recombinants, all subtype F specimens, and eight selected subtype B samples were further evaluated by sequence analysis, which confirmed the results of the screening assays (Figure 2a, 2b).
The search for the HIV-1 recombinant genome in these 18 samples was further expanded to the gag region. Mosaic sequences of subtypes B and F were found within the p17 or p24 gag (Table 2, Figure 2, discussion below) in four prt-env subtype F variants (BR46, BR57, BR59, and BR97). Similarly, in one of two prt-env recombinants (BR60), the gag sequences had a mosaic pattern. In contrast, the p24 gag sequences of eight prt-env subtype B specimens were homogeneous and also classified as subtype B (Figure 2). Taken together, the comparative molecular analysis of gag, pol, and env regions allowed the identification of six specimens that carried HIV-1 recombinant genomes, representing five distinct mosaic structures: Bgag-p17/Bgag -p24/Fpol/Benv , Fgag-p17/Bgag -p24/Fpol/Fenv , Bgag-p17/B-Fgag -p24/Fpol/Fenv , Bgag-p17/B-Fgag -p24/Fpol/Benv , and Fgag-p17/B-Fgag -p24/Fpol/Fenv .
| Table 2. p17/p24 gag, prt, and C2-V3 genetic subtyping of HIV-1 DNA sequencesa from peripheral blood mononuclear cells collected from 18 patients in Rio de Janeiro | |||||
| Genotypes | |||||
| Specimen | No. | gag p17 | gag p24 | prt | C2-V3 |
| Group 1b | 8 | NDc | B | B | B |
| Group 2d | 4 | F | F | F | F |
| (BR46) | (NAe) | (B) | (F) | (F) | |
| (BR59) | (F) | (B) | (F) | (F) | |
| (BR57) | (F) | (B/F) | (F) | (F) | |
| (BR97) | (B) | (B/F) | (F) | (F) | |
| (BR60) | (B) | (B/F) | (F) | (B) | |
| (BR43) | (B) | (B) | (F) | (B) | |
| aRecombinant specimens are shown in parentheses; B/F
mosaic structure within a 311-bp of the p24 gag fragment consisting of subtypes B
and F sequences. bGroup 1: BR34, BR52, BR55, BR64, BR65, BR71, BR75, and BR92. cND=not done. dGroup 2: BR41, BR54, BR58, and BR112. eNA = not available due to negative PCR. |
|||||
 |
|
Click here to view enlarged image
|
The potential crossover breakpoints within 717-bp of the p17-p24 gag mosaic
sequences were examined by comparison with nucleotide signature patterns characteristic
for subtypes B and F viruses (Figure 3). We performed comparative analyses with aligned
DNA sequences of recombinants (BR57, BR59, BR60, and BR97), subtype B (MN and BR43), and
subtype F variants (BR41, BR54, BR58, and BR112). This analysis confirmed an intragene
recombination within p24 gag in specimens BR57, BR60, and BR97—a finding
consistent with our failure to phylogenetically assign these gag sequences to any
known subtype (Figure 2c).
The putative breakpoints within the intragene recombinant sequences were located between nucleotides 97 and 137 in sample BR57 and between nucleotides 173 and 213 in BR60 and BR97 (Figure 3). The exact breakpoint position could not be determined because of extensive sequence homology between subtypes B and F in this viral region. This analysis also revealed putative crossover breakpoints for variants BR57 and BR59 in proximity to the coding region for the p17-p24 protein-processing site. The potential breakpoints between the second half of gag and the beginning of pol region in variants BR43, BR46, and BR59 are being investigated.
To examine the possibility of in vitro recombination during PCR amplification (30), we performed PCR amplification of the long fragments covering the gag and prt area in the endpoint- diluted lysates (31). To ensure that endpoint PCR products were amplified from single copy templates, we used samples only from dilutions at which 1 of 10 PCR amplifications were productive for further sequence analysis. The comparative analysis of the entire p17 gag, p24 gag, and prt sequences demonstrated 98% homology between the undiluted and diluted lysates—strong evidence that the recombinant sequences were not a result of the PCR amplification process.
Epidemiologic and Clinical Characteristics
The mean ages of patients infected with HIV-1 of single subtype B or F were not different from those with dual or recombinant infections (p = 0.77) (Table 3). In addition, the patients did not differ significantly by gender, risk group, and clinical stage of disease (p = 0.44 to p = 0.48). The patients infected with HIV-1 subtype B (403) and subtype F (854) did, however, differ (p = 0.04) by mean CD4 counts. Although dates of seroconversion were not known for all patients, the earliest HIV-positive results were reported among patients infected with subtype B (in agreement with previous observations that the spread of subtype B viruses occurred earlier than other HIV-1 subtypes in Brazil [16,17]), which might explain the difference in CD4 counts.
Table 3. Characteristics of the study population |
||||
| Characteristic | Dual infection n=3 |
Recombinant infection n=6 |
Subtype B n=66 |
Subtype F n=4 |
| Gender (Female/male) | 1/2 | 3/3 | 24/42 | 3/1 |
| Mean age in years (range) | 36 (30-44) | 34 (25-45) | 37 (23-60) | 34 (26-43) |
| Clinical stagea | ||||
|
3 | 5 | 29 | 4 |
|
– | 1 | 17 | – |
|
– | – | 14 | – |
|
– | – | 6 | – |
| Mean CD4 cells (range) | 527 (404-743) | 484 (190-821) | 403 (59-1281)b | 823 (570-1270) |
| Years of 1st serologic tests | ||||
|
– | – | 2 | – |
|
2 | 2 | 30 | 2 |
|
1 | 4 | 34 | 2 |
| Heterosexual | 2 | 5 | 27 | 4 |
| Homosexual | 1 | – | 19 | – |
| Bisexual | – | – | 8 | – |
| Blood transfusion recipient | – | – | 3 | – |
| Intravenous drug user | – | – | 1 | – |
| Multiple factors | – | 1 | 2 | – |
| Unknown | – | – | 6 | – |
| aWHO staging system [32]. bBased on data available for 54 patients. |
||||
Conclusions
HIV-1 infections caused by dual and intersubtype-recombinant genome may be relatively common among HIV-1-infected Brazilians. Using both heteroduplex mobility assay and RFLP screening methods, as well as sequencing, we identified three dual (3.8%) and six recombinant (7.6%) infections involving distinct viral subtypes among 79 HIV-1-infected persons from Rio de Janeiro.
We chose viral prt to screen for dual infections because this highly conserved region provides the best opportunity for simultaneous amplification of distinct HIV-1 variants in the same PCR reaction. Proviral prt sequences can be routinely amplified from approximately 95% of all analyzed seropositive samples collected from the Americas, Asia, Africa, and Europe (data not shown). Moreover, the RFLP assay of the prt gene is convenient for screening a large number of samples (5). The detection of B/F, B/D, and F/D dual infections is in agreement with our 1993 study among patients from the same Rio de Janeiro cohort, which showed five dual HIV-1 infections involving subtypes F and B (one case), F and D (one case), and B and C (three cases of familial clustering) among 21 HIV-infected persons (5-7). Interestingly, dual infections involving HIV-1 subtype D continue to be detected in patients from Rio de Janeiro, an area with a high predominance of HIV-1 subtypes B and F, but rare subtype D, single infections. Because retrospective specimens (peripheral blood mononuclear cells) were not available for these patients, we could not confirm whether the acquisition of mixed strains was sequential (superinfection) or simultaneous. However, combination of subtypes F or B with rare subtype D viruses among some dual infections may suggest that in these cases two viral strains might be acquired through cotransmission rather than through superinfection.
All naturally occurring HIV-1 dual infections are likely not detected by current methods. First, quantitative differences in two distinct viral DNA templates in the sample can lead to selective PCR amplification of only one subtype. Second, despite targeting conserved genes such as prt, some divergent viral strains may escape PCR amplification because of primer mismatches. Finally, a single nucleotide mutation in the endonuclease restriction site can abrogate the recognition pattern and distort detection of dual infections in the RFLP analysis. These observations suggest that the rate of HIV-1 mixed infections within this Brazilian cohort might be even higher than 4%. Our previous findings of five dual infections among 21 patients from the same cohort support this assumption. If we take into account these five cases, the percentage of mixed infections caused by viruses of distinct subtypes circulating between 1993 and 1994 among 100 patients analyzed from this Rio de Janeiro cohort would increase to 8%.
The potential underestimate of mixed infections is highlighted by the additional detection of 6 (7.6%) distinct recombinants within the cohort. This finding is consistent with the estimated 5% to 10% intersubtype mosaics among HIV-1 genomes in the Los Alamos database (3). Interestingly, all recombinant or mosaic genomes described in this report involved only subtypes B and F viral regions, although dual infections caused by other subtypes were also circulating in this cohort. Moreover, our results indicate that recombination between gag and pol (prt) regions is more frequent than between pol and env and lead to speculation that such stable B/F mosaics have selective advantage. Such observations support the assumption that recombination within the gag gene occurs more often than within other viral regions (33) and emphasize the need for rapid subtyping methods specific to the gag region.
To investigate the potential impact of dual and recombinant infections on the clinical status of patients, we compared the clinical and demographic characteristics of these patients with those of patients infected with one nonrecombinant viral subtype. Epidemiologic information for all 79 patients was available only at the first draw of blood. Although the results did not show significant differences between the two groups, the possibility of differences exists.
Our findings provide the first baseline measure of the range of HIV variability in Rio de Janeiro from 1993 to 1994 and the proportion of dual and recombinant infections among the HIV-infected Brazilian population. Future systematic molecular epidemiologic surveys of HIV heterogeneity in Rio de Janeiro may show the potential changes in the molecular profile of the HIV/AIDS epidemic over time; the laboratory tools we used to identify single, dual, and recombinant infections may be useful in such investigations. Nevertheless, our study on genetic variation of HIV-1 subtypes among blood donors from the state of Rio de Janeiro documented the presence of mosaic viruses of subtypes B and F and subtypes B and D in blood units collected in 1996 (34). Moreover, recent genetic analysis of viral strains collected in 1997 from HIV-1-infected patients living in Manaus (a city in Brazil's Amazon region) showed the presence of dual infections and recombinants caused by subtypes B and F viruses (A. Tanuri, pers. comm.). Therefore, these data indicate that the heterogenic pattern of HIV-1 infections, first observed in Rio de Janeiro, also exists in other regions of Brazil.
Our findings indicate that mixed infections and mosaic viruses may be more common in worldwide epidemics than previously thought and, therefore, may provide a basis for developing AIDS vaccines and predicting the global evolution of HIV.
Acknowledgments
We thank Priscilla Swanson for her help in sequencing and Renu Lal for critical review of the manuscript.
Artur Ramos is a graduate fellow at the Federal University of Rio de Janeiro, Rio de Janeiro, Brazil. His laboratory research focuses on HIV-1 genetic variability worldwide. His research interests include application of molecular biology techniques to epidemiology of HIV.
Address for correspondence: Danuta Pieniazek, HIV/Retrovirus Diseases Branch, Division of AIDS, STD, and TB Laboratory Research, CDC, 1600 Clifton Road, MS G19, Atlanta, GA, 30333, USA; fax: 404-639-1010; e-mail: dxp1@cdc.gov.
References
![]()
Top of Page | Current
Issue | Upcoming Issue | Past Issue | Search
| Home