Research
Nancy Hoe,* Kazumitsu Nakashima,* Diana Grigsby,* Xi Pan,* Shu Jun Dou,* Steven
Naidich,† Marianne Garcia,‡ Emily Kahn,‡ David Bergmire-Sweat,‡ and
James M. Musser*
*Baylor College of Medicine, Houston, Texas, USA; †Naidich Space Laboratory, Inc.,
New York, New York, USA; ‡Texas Department of Health, Austin, Texas, USA
| Serotype M1 group A Streptococcus, the most common cause of invasive disease in many case series, generally have resisted extensive molecular subtyping by standard techniques (e.g., multilocus enzyme electrophoresis, pulsed-field gel electrophoresis). We used automated sequencing of the sic gene encoding streptococcal inhibitor of complement and of a region of the chromosome with direct repeat sequences to unambiguously differentiate 30 M1 isolates recovered from 28 patients in Texas with invasive disease episodes temporally clustered and thought to represent an outbreak. Sequencing of the emm gene was less useful for M1 strain differentiation, and restriction fragment length polymorphism analysis with IS1548 or IS1562 as Southern hybridization probes did not provide epidemiologically useful subtyping information. Sequence polymorphism in the direct repeat region of the chromosome and IS1548 profiling data support the hypothesis that M1 organisms have two main evolutionary lineages marked by the presence or absence of the speA2 allele encoding streptococcal pyrogenic exotoxin A2. |
Molecular genetic approaches that differentiate isolates of a pathogenic microbial species have revolutionized contemporary epidemiologic investigations of putative disease outbreaks. The human gram-positive bacterium group A Streptococcus (GAS) has more than 80 M-protein serotypes, but isolates expressing the M1 serotype are disproportionately represented among invasive disease episodes in most case series (1). M1 organisms also commonly cause pharyngitis. For reasons that are unknown, M1 isolates and organisms expressing other M serologic types can undergo rapid temporal variation in disease frequency and severity (1). Serotype M1 isolates have been studied by several molecular typing approaches, including multilocus enzyme electrophoresis; pulsed-field gel electrophoresis; rRNA gene polymorphism typing (ribotyping); random amplified polymorphic DNA analysis; and sequencing of the genes encoding streptokinase, C5a peptidase, M protein, hyaluronidase, and pyrogenic exotoxin A, B, and C (1-5). The common theme of these analyses is that most M1 isolates cultured from patients with invasive disease episodes are closely allied in overall chromosomal relationship as a consequence of sharing a recent common ancestor (1,3,5). Lack of readily detectable chromosomal variation has limited insights on the molecular origin of new virulent strains, velocity of strain spread in human populations, and association of genetic subtypes with certain clinical syndromes, including necrotizing fasciitis and acute rheumatic fever.
Recently, Akesson et al. (6) identified a GAS extracellular protein made by M1 strains that inhibits human complement. This streptococcal inhibitor of complement (Sic) protein is incorporated into the membrane-attack complex (C5b-C9) and inhibits target cell lysis by an undetermined mechanism. Analysis of molecular diversity among 16 M1 GAS isolates from patients with pharyngitis identified seven alleles of the sic gene (7). The high level of sic polymorphism was unanticipated, given that other methods of molecular analysis had failed to identify substantial variation among M1 isolates (1-5). Subsequently, Stockbauer et al. (8) analyzed 165 M1 isolates from diverse localities, identified 62 alleles, and documented a uniquely high level of allelic variation in this gene. The molecular features of sic variation indicated that structural change in Sic is mediated by natural selection (8). Moreover, study of 70 M1 isolates from two temporally distinct epidemics of streptococcal infections in the former East Germany suggested that variation in sic contributed to fluctuations in GAS disease frequency and severity (8).
The observation that the polymorphism in the sic gene greatly exceeded that for all other genes examined in serotype M1 isolates suggested that sic sequencing could be used as a rapid strategy to differentiate organisms thought to be epidemiologically linked. A recent statistically significant increase in cases of invasive GAS in Texas presented an opportunity to test this hypothesis. We also tested whether molecular variation in a region of the chromosome with multiple direct repeat (DR) nucleotide sequences and restriction fragment length polymorphism (RFLP) analysis with insertion elements IS1548 (9) and IS1562 (10) would differentiate M1 isolates.
Statistics gathered by the Texas Department of Health indicated that from December 1, 1997, through March 5, 1998, 117 invasive episodes of GAS (and 26 deaths) had occurred statewide. Sixty of these cases and 14 deaths were in central Texas (population 1.4 million). Concern was raised by community physicians, lay individuals, and the media that an unusually virulent strain was causing a disease outbreak. (A complete description of the epidemiology of this outbreak will be presented elsewhere.) For molecular analysis of the GAS causing recent cases, 100 isolates were sent to the laboratory of J.M.M. at Baylor College of Medicine, Houston, TX. On receipt, the bacteria were checked for purity by visual inspection and were confirmed to contain beta-hemolytic organisms with a colony morphology consistent with GAS. Chromosomal DNA was isolated as described (5).
To determine whether one or a few unusually virulent strains might account for most of the invasive episodes, we sequenced the hypervariable part of the emm gene encoding M-type specificity (5,11). After the sequence data were edited electronically, they were used to search an emm database maintained in the laboratory that contains at least one sequence of all known M-protein serotypes and provisional serotypes (11). The database also contains 33 emm1 allelic variants identified among serotype M1 organisms from global sources (1,5,12) (Figure 1).
 |
Click here to view enlarged imageFigure 1. Alignment of inferred N-terminal amino acid sequences of 33 alleles of emm1. The region shown represents amino acids 27 through 110 (GenBank accession number X07860). Six of the emm1 alleles were identified in this study, several were described previously (1,5,12), and others were from ongoing analysis of emm1 in M1 strains from global sources. Amino acid residues identical to those encoded by emm1.0 are represented by periods. |
The most common M type identified was M1 (n = 30 isolates) (Table). Five emm1 alleles were identified in the 30 M1 isolates, including four (emm1.13, emm1.18, emm1.19, and emm1.24) not previously described (Figure 1). Twenty-three Texas isolates had allele emm1.0, the most common emm1 allele in M1 isolates globally (5). Three isolates had allele emm1.19, two organisms had allele emm1.24, and one isolate each had allele emm1.13 and emm1.18 (Table). Compared with the emm1.0 allele encoding variant M1.0, each of these alleles is characterized by single nucleotide changes resulting in single amino acid substitutions in the resulting M1 protein (Figure 1). The additional 70 isolates were a heterogeneous array of M types, including M3, M4, M5, M6, M12, M18, and many others. A more detailed description of the bacteriologic features will be presented elsewhere.
Because M1 isolates were a prominent cause of the invasive disease episodes, we sought to determine the extent of genotypic heterogeneity among the 30 M1 GAS isolates. First, polymerase chain reaction (PCR) was used to test whether the organisms possessed the speA gene encoding pyrogenic exotoxin A (scarlet fever toxin) (3,13). Most contemporary M1 isolates cultured from patients with invasive disease have this gene (1,3-5), but some lack it because speA is bacteriophage encoded (13). Possession of speA is therefore a variable trait among M1 organisms. All 30 M1 isolates had the speA gene, and sequence analysis of 11 random isolates found that all had allele speA2 (14). Previous study of the speA gene in several hundred contemporary M1 strains showed that all organisms had the speA2 allele (1,14).
| Table. Characteristics of serotype M1 Group A Streptococcus isolates analyzed | |||||||
| MGAS no.a |
TDH no.b | sic allele |
emm1 allele |
DRc PCRd (bp) |
DR se- quence type |
speA PCRe |
IS1548 type |
| 6151 | BE8-776 | 1.01 | 1.0 | 372 | 4.0 | pos | 1.0 |
| 6168 | BE-98-743 | 1.01 | 1.0 | 306 | 3.0 | pos | 1.0 |
| 6184 | BE8-873 | 1.01 | 1.0 | 306 | NSf | pos | 1.0 |
| 6199 | BE8-917 | 1.01 | 1.19 | 306 | NS | pos | 1.0 |
| 6262 | BE8-1085 | 1.01 | 1.19 | 306 | NS | pos | 1.0 |
| 6264 | BE8-1087 | 1.01 | 1.19 | 306 | 3.0 | pos | 1.0 |
| 6181 | BE-98-764 | 1.02 | 1.0 | 240 | NS | pos | 1.0 |
| 6293 | BE8-1339 | 1.02 | 1.0 | 306 | NS | pos | 1.0 |
| 6294 | BE8-1340 | 1.02 | 1.0 | 306 | NS | pos | 1.4 |
| 6140 | BE8-629 | 1.13 | 1.0 | 240 | NS | pos | 1.0 |
| 6200 | BE8-918 | 1.13 | 1.0 | 240 | NS | pos | 1.0 |
| 6201 | BE8-919 | 1.13 | 1.0 | 240 | NS | pos | 1.0 |
| 6281 | BE8-1149 | 1.13 | 1.24 | 306 | NS | pos | 1.3 |
| 6137 | BE8-563 | 1.32 | 1.0 | 306 | 3.0 | pos | 1.0 |
| 6148 | BE8-773 | 1.32 | 1.0 | 306 | NS | pos | 1.0 |
| 6249 | BE8-929 | 1.32 | 1.0 | 306 | NS | pos | 1.0 |
| 6172 | BE-98-751 | 1.34 | 1.0 | 306 | NS | pos | 1.0 |
| 5997 | BE8-191 | 1.36 | 1.0 | 240 | NS | pos | 1.0 |
| 6135 | BE8-548 | 1.36 | 1.0 | 240 | 2.2 | pos | 1.0 |
| 6254 | BE8-1021 | 1.36 | 1.24 | 306 | NS | pos | 1.0 |
| 6189 | BE8-88 | 1.66 | 1.13 | 306 | NS | pos | 1.0 |
| 5999 | BE8-208 | 1.99 | 1.0 | 306 | 3.0 | pos | 1.0 |
| 6003 | BE8-322 | 1.100 | 1.0 | 240 | NS | pos | 1.0 |
| 6251 | BE8-1000 | 1.100 | 1.0 | 240 | 2.1 | pos | 1.0 |
| 6006 | BE8-369 | 1.101 | 1.0 | 306 | 3.0 | pos | 1.0 |
| 6138 | BE8-566 | 1.118 | 1.0 | 240 | 2.2 | pos | 1.0 |
| 6150 | BE8-775 | 1.119 | 1.0 | 306 | 3.0 | pos | 1.0 |
| 6154 | BE8-792 | 1.120 | 1.18 | 240 | 2.1 | pos | 1.0 |
| 6272 | BE8-1111 | 1.179 | 1.0 | 306 | NS | pos | 1.0 |
| 6299 | BE8-1380 | 1.180 | 1.0 | 240 | 2.0 | pos | 1.0 |
| 2221 | NA | 1.01 | 1.0 | 306 | NS | pos | 1.0 |
| 5305 | NA | 1.01 | 1.0 | 306 | 3.0 | pos | 1.0 |
| 5809 | NA | 1.01 | 1.0 | 305 | 3.01 | pos | 1.0 |
| 2139 | NA | 1.02 | 1.0 | 306 | 3.0 | pos | 1.0 |
| 2350 | NA | 1.09 | 1.0 | 306 | 3.0 | pos | 1.0 |
| 1272 | NA | 1.35 | 1.0 | 306 | NS | pos | 1.5 |
| 5297 | NA | 1.121 | 1.0 | 240 | 2.0 | pos | 1.0 |
| 279 | NA | 1.08 | 1.3 | 570 | 7.0 | neg | 1.6 |
| 1632 | NA | 1.08 | 1.3 | 570 | 7.0 | neg | 1.6 |
| 1653 | NA | 1.19 | 1.3 | 570 | 7.0 | neg | 1.6 |
| 326 | NA | 1.20 | 1.3 | 570 | 7.0 | neg | 1.6 |
| 570 | NA | 1.21 | 1.3 | 570 | 7.0 | neg | 1.8 |
| 1642 | NA | 1.24 | 1.3 | 504 | 6.1 | neg | 1.6 |
| 6708g | NA | 1.225 | 1.6 | 504 | 6.0 | neg | 1.7 |
| aMGAS, Musser group A Streptococcus
strain number. All isolates had no known direct epidemiologic connection except MGAS 6199,
6264, and 6272 (associated household cases); MGAS 6140, 6200, and 6201 (blood and
cerebrospinal fluid cultures of same patient); and MGAS 6293 and 6294 (mother-neonate
paired isolates). bTDH, Texas Department of Health strain number; NA, not applicable (control isolate). cDR, direct repeat. dPCR, polymerase chain reaction. epos, PCR-positive for speA; neg, PCR-negative for speA. The speA gene in MGAS 1272, 6135, 6137, 6138, 6150, 6151, 6154, 6168, 6251, 6264, 6272, and 6299 was sequenced and identified as allele speA2. fNS, not sequenced. gMGAS 6708 is also known as SF370. The genome of this organism is being sequenced at the University of Oklahoma. |
|||||||
Recent molecular genetic studies have documented that sic is a uniquely hypervariable gene among M1 GAS strains (7,8). Our sic database consists of 252 distinct alleles identified by sequence analysis of ~1,200 M1 isolates from worldwide sources and cultured from patients with a large array of GAS diseases, including pharyngitis and invasive episodes (7;8; unpub. data). sic allelic variation has not been identified during in vitro laboratory passage, nor has variation been detected among strains that are epidemiologically associated (8). These molecular features suggest that automated sequencing of sic may be a convenient method for identifying M1 genetic subtypes and inferring epidemiologic relationships in potential outbreaks. To test this idea, we sequenced the sic gene in the 30 M1 isolates and identified 15 sic alleles that differed from one another by at least one nucleotide (Figure 2). Seven of the 15 alleles were not found among the ~1,200 M1 isolates previously characterized for sic variation. Eight new nucleotide substitutions were identified in eight codons, and one codon had a new dinucleotide change; these changes would result in nine amino acid substitutions in the expressed Sic proteins. As observed in earlier analyses (7,8), the amino-terminal half of the Sic protein had many insertions and deletions, all in frame (Figure 2).
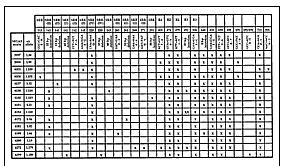 |
Click here to view enlarged imageFigure 2. Variation in the sic gene and Sic protein identified in M1 group A Streptococcus isolates characterized in the study. The figure is a compilation of variations found in the 15 distinct sic alleles in the sample. The numbers at the top of the figure refer to the nucleotide sequence position of a sic allele described in reference 6. Single-letter amino acid abbreviations are used. SRR, amino-terminal short repeat region; Roman numeral, short repeats I-V which recur in SRR; R2 and R3, tandem repeats; MGAS strain, Musser Group A Streptococcus strain number; X, presence of polymorphism. |
IS1548, a recently described insertion sequence, has been reported to be polymorphic in copy number and location in the chromosome of group A and group B streptococci (9). IS1562 is an insertion sequence located in the Mga regulon between the sic gene and scpA gene encoding C5a peptidase in some GAS (10). Relatively few GAS strains have been analyzed by RFLP profiling with these elements, and their ability to differentiate among isolates expressing the same M type has not been assessed. Since insertion sequence profiling has helped elucidate transmission dynamics and evolutionary relationships of Mycobacterium tuberculosis (15), Bordetella pertussis (16), Streptococcus pneumoniae (17), Escherichia coli (18), and Salmonella Enteritidis (19), we tested the hypothesis that IS1548 or IS1562 subtyping would provide additional epidemiologically informative data regarding genetic diversity among M1 isolates.
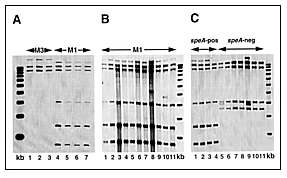
To determine whether the IS1548 element was present in M1 organisms in our sample, PCR was performed on genomic DNA from 10 random isolates by using the oligonucleotides (forward) 5'-TGCCGTTCATCAACTGATTTCAGTGG-3' and (reverse 5'-CGACGATAACTGAGGTCTTTTTT AGGAAAT-3'(9). A PCR product of the anticipated size of ~1 kb was obtained from all organisms, a result indicating that the isolates had this element or a close relative. The PCR-amplified fragment was subsequently used as a probe for RFLP analysis by Southern blotting after EcoNI digestion and electrophoretic separation of chromosomal DNA fragments. The data were analyzed with a Bioimage Analyzer system interfaced with a Sun Sparcstation. Four M1 isolates had the same 6-band IS1548 RFLP pattern, which was distinct from the 3-band pattern obtained from three random serotype M3 isolates (Figure 3A). Twenty-eight of the 30 M1 isolates studied had the same IS1548 pattern (Figure 3B and data not shown). The IS1548 RFLP patterns of the two other isolates were single-band variants of the common M1 pattern, both characterized by the addition of one hybridizing band (Figure 3B). One of the isolates (MGAS 6294) with a variant IS1548 pattern was recovered from the blood of a neonate born to a woman with GAS sepsis. The isolate (MGAS 6293) from the blood of the infected mother had the common IS1548 pattern.
 |
|
Click here to view enlarged imageFigure 3. Representative IS1548 RFLP fingerprint patterns of M1 isolates. Panel A is a lane map showing results from analysis of three serotype M3 control isolates and four M1 isolates with different sic alleles. Lane 1, MGAS5892; lane 2, MGAS6004; lane 3, MGAS6005; lane 4, MGAS5997; lane 5, MGAS5999; lane 6, MGAS6003; lane 7, MGAS6006. kb, 1-kb DNA ladder. Panel B is a lane map showing results from analysis of eleven M1 isolates with eight different sic alleles. Lane 1, MGAS6201; lane 2, MGAS6249; lane 3, MGAS6251; lane 4, MGAS6254; lane 5, MGAS6262; lane 6, MGAS6264; lane 7, MGAS6272; lane 8, MGAS6281; lane 9, MGAS6293; lane 10, MGAS6294; lane 11, MGAS6299. kb, 1-kb DNA ladder. Panel C is a lane map showing results from analysis of four speA-positive and seven speA-negative M1 isolates. Lane 1, MGAS2350, lane 2, MGAS2221, lane 3, MGAS2139, lane 4, MGAS1272, lane 5, MGAS6708, lane 6, MGAS1653, lane 7, MGAS1642, lane 8, MGAS1632, lane 9, MGAS570, lane 10, MGAS326, lane 11, MGAS279. kb, 1-kb DNA ladder. |
To identify other IS1548 RFLP patterns in M1 GAS organisms, we analyzed 14 non-Texas control isolates. These 14 M1 isolates were selected for analysis because they have been well characterized by several molecular techniques (5). The isolates also have many different sic alleles and include representatives of two major genetic subclones of M1 organisms (5). IS1548 profiling of this group identified the common six-band pattern and also found five organisms with a distinct subtype with four bands (Figure 3C). All organisms with this profile were speA-negative. Interestingly, MGAS6708 (SF370), the M1 strain whose genome is being sequenced (20), had a unique five-band IS1548 fingerprint (Figure 3C). The IS1548 profile for this strain was very similar to the four-copy pattern characteristic of most of the speA negative organisms.
We next used PCR to determine whether IS1562 was present in the 30 M1 organisms from Texas and in 11 of the 14 non-Texas isolates by using oligonucleotide primers 3244 and 3267, as described by Berge et al. (10). A PCR product of the expected size of ~1 kb was obtained from all isolates. The ~1-kb fragment was used to reprobe the nylon membranes used for IS1548 RFLP analysis. The results showed that all M1 isolates tested had the identical or closely similar RFLP characterized by one copy of IS1562 (data not shown).
Several years ago Groenen et al. (21) characterized an unusual region of the M. tuberculosis chromosome that contains up to approximately 40 copies of a 36-bp DR sequence interspersed with unique-sequence spacer regions 35 bp to 41 bp in length. Subsequent analysis of this DR region in hundreds of M. tuberculosis isolates by a method referred to as spacer oligotyping (spoligotyping) has identified large numbers of distinct subtypes of this pathogen (22), indicating that the DR region is highly polymorphic, even among isolates closely related in overall chromosomal character (23). We examined the M1 GAS genome database maintained by the University of Oklahoma Advanced Center for Genome Technology and identified a region of the GAS chromosome located on contig 208 (database as of February 22, 1999) that consists of seven DR elements separated by six unique 30-bp spacer regions. This area of the M1 chromosome is referred to as a DR region on the basis of its shared structural features with the M. tuberculosis DR region.
To test the hypothesis that the DR region is polymorphic among M1 GAS isolates, we analyzed the 14 control isolates by PCR with primers that flank this region (DR003, 5'-GGGCTTTTCAAGACTGAAGTCTAGCTG-3' and DR004, 5'-TCCGACTGCTGGTATTAACCCTC TT-3'). Four sizes of PCR products were identified (data not shown). Six of seven isolates previously identified as RFLP type 1a (speA-positive, containing allele emm1.0) had an apparently identical size PCR product of ~300 bp. A PCR product of ~240 bp was identified in the remaining isolate. Two sizes of PCR products (~500 bp and ~570 bp) were also identified in the six organisms with RFLP type 1k (speA-negative, allele emm1.3). Hence, the PCR results indicated that size variation was present in the GAS DR region in M1 organisms and showed that isolates of the RFLP types 1a and 1k categories did not share PCR fragment sizes.
To examine nucleotide variation in this chromosomal region, we sequenced the PCR products obtained from 12 of these control M1 isolates, including 5 with the ~240-bp or ~300-bp PCR product and 7 organisms with either the ~500-bp or ~570-bp PCR product. The one organism with the ~240-bp PCR product, characterized by two identical DR elements and two nonidentical spacer sequences, is arbitrarily designated DR type 2.0 (Figure 4). Three of the four organisms with the ~300-bp PCR product had identical DR-region sequences defined by the presence of three identical DR elements and three nonidentical spacer sequences (Figure 4B). This molecular arrangement was designated DR type 3.0 (Figure 4C). The DR element of the fourth isolate differed from the other three by the absence of 1 base in the second spacer region and is designated DR type 3.01 (Figure 4C). Consistent with the difference in PCR fragment size, the sequences of the DR region in the seven other organisms were distinct from the DR type 3.0 sequence. Five of these seven isolates had an identical DR-region sequence that was characterized by seven spacer regions (designated DR type 7.0). Two organisms lacked one of the spacer regions present in the DR type 7.0 strains; these molecular variants were designated DR types 6.0 and 6.1 (Figure 4C).
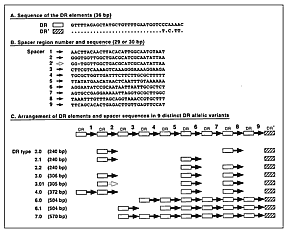 |
Click here to view enlarged imageFigure 4. Polymorphism identified in the direct repeat (DR) region of serotype M1 group A Streptococcus. The data were generated by automated DNA sequencing of polymerase chain reaction products obtained with the oligonucleotide primers DR003 and DR004 described in the text. (A) The 36-bp sequences of the two related DR and DR' elements. Multiple copies of the DR element present in different M1 isolates all had the identical sequence. (B) The 29-bp or 30-bp sequences of the 10 distinct spacer regions identified in the analysis. (C) Arrangement of the DR elements and spacer sequences in nine distinct DR allelic variants. The DR types were given arbitrary designations based in part on the number of DR elements present. Open or cross-hatched rectangles represent copies of the DR or DR' elements; arrows represent copies of the spacer region sequences connecting the DR elements. The numbers above the spacer region sequences refer to the spacers designated in part B of the figure. |
We next analyzed the 30 M1 Texas isolates by PCR of the DR region and obtained three PCR fragment sizes: products of ~240 bp (n = 11 isolates), ~300 bp (n = 18 isolates), and ~370 bp (n = 1 isolate). We sequenced the PCR products from 12 organisms selected to represent an array of DR PCR fragment sizes and emm and sic alleles. Two additional sequences (designated DR types 2.1 and 2.2) were identified among the five isolates with the DR region PCR fragment size of ~240 bp. All six isolates with the ~300-bp PCR product had the identical sequence (DR type 3.0). The one isolate with the ~370-bp PCR product had a unique sequence (DR type 4.0) with four spacer regions (Figure 4). The results showed that the DR region had more molecular variation than emm. However, the level of allelic variation in sic exceeded that found in either emm or the DR region.
Our data underscore the importance of molecular typing techniques in rapidly providing information about the epidemiology of GAS infections (24). The emm sequence data indicated that a heterogeneous array of GAS M types was present in the sample of 100 GAS isolates; thus, we could rapidly rule out the notion that the invasive cases had been caused by one or a few distinct GAS strains. Moreover, molecular analysis of several other polymorphic loci, including automated DNA sequencing of sic and a chromosomal region with multiple DR sequences, showed that M1 organisms, the most abundant serotype in the sample, had substantial levels of genetic diversity. Of the molecular techniques used in this analysis, sequencing the sic gene was the most effective for differentiating among M1 isolates because it identified the most variants. RFLP-based typing with IS1548 and IS1562 failed to provide extensive, or even adequate, resolving power among the M1 organisms for epidemiologic purposes. Moreover, the variation in the IS1548 RFLP profile we detected in two isolates (MGAS 6293 and MGAS 6294) from a woman with puerperal sepsis and the blood of her newborn child suggests that IS1548 can be mobile in host-pathogen interactions. Instability in insertion sequence profiles has also been reported for IS6110, an element commonly used for molecular subtyping of M. tuberculosis (25).
Although sequence analysis of emm and the DR region provided some useful molecular subtyping data for M1 strains, the level of polymorphism at these loci was less than in sic. A rapid PCR-based subtyping system to index polymorphism in the DR region could be formulated for M1 GAS that would be similar to the method available for M. tuberculosis. However, this approach would be less useful for M1 GAS than M. tuberculosis because in the latter organism 43 distinct spacer regions have been described. Hence, the number of polymorphic markers is considerably greater than in M1 GAS, in which thus far only 13 spacer regions have been found (unpub. data).
Our work, recently reported results (7,8), and unpublished data obtained from ongoing analysis of sic polymorphism in large samples obtained from population-based studies demonstrate four emerging themes in the molecular epidemiology and evolutionary biology of M1 organisms. First, several sic variants are dispersed over broad geographic areas; some have achieved intercontinental distribution. For example, M1 strains with the sic1.01 allele have been identified in 14 countries. This allele might be widely disseminated because it is the ancestral condition in M1 organisms or otherwise has had a long-standing association with the M1 serotype. Another plausible hypothesis to explain its widespread dissemination is that expression of Sic1.01 protein bestows greater fitness than do other Sic variants. A third possibility is that the Sic1.01 variant marks an M1 subclone with an unusual propensity to survive and spread. In this regard, we note that virtually all isolates with the sic1.01 allele are speA-positive. GAS isolates with the speA gene are statistically overrepresented among organisms recovered from children with pharyngitis who have not been cured by oral antibiotic therapy (26). Bacterial survival despite appropriate antibiotic therapy would likely enhance spread of the organism to new hosts and, hence, assist widespread dispersal. We also note that speA-positive M1 isolates are internalized efficiently by human respiratory tract epithelial cells grown in culture (27,28), a process that could provide access to a protective niche that enhances survival capability.
A second important theme is that many sic alleles are confined to local geographic areas (e.g., individual countries or communities). For example, seven of the sic alleles identified in this study were unique to the Texas M1 isolates. Several unique sic alleles also were found among organisms cultured from patients in Mexico (7) and the former East Germany (8). Because many sic alleles can be readily linked with one another by a single molecular event such as a nucleotide substitution or one insertion or deletion, some of the variants likely arise rapidly in local areas. Their absence in other regions is explained by lack of sufficient elapsed time required for widespread dispersal. Recent data obtained from study of M1 isolates recovered from population-based surveys in Finland (29), Ontario, Canada (30), and Atlanta, Georgia (31) strongly support this explanation (unpub. data).
The third theme is the remarkable polymorphism in the sic gene. Stockbauer et al. (8) reported that virtually all changes in the sic gene result in structural changes in the Sic protein and concluded that positive Darwinian selection is mediating Sic variation. Our study confirmed these observations. For example, all 10 new nucleotide changes identified would result in amino acid substitutions in Sic, and all insertions and deletions were in frame. Moreover, most of the amino acid changes were radical replacements, that is, those producing charge changes or polar-nonpolar substitutions. These types of amino acid replacements commonly result in functional differences in the resulting proteins and are a hallmark of positive selection (32).
Last, accumulating data suggest the existence of two genetically divergent M1 subpopulations, which can be thought of as two evolutionarily distinct lineages. Our study found that organisms with the speA gene and chromosomal PFGE type 1a (5) have shorter DR-region sequences and an IS1548 profile characterized by six hybridizing bands. In contrast, organisms that are speA-negative usually have PFGE type 1k (5), longer DR sequences, and an IS1548 fingerprint with four bands. In addition, we will show elsewhere that the two M1 lineages each have distinct families of sic alleles. Together, the data indicate that sufficient time has elapsed since a shared common ancestor for members of the two lineages to have diverged at many chromosomal loci. The data also indicate that transduction of the speA2 allele between members of the two lineages is apparently rare in natural populations of GAS (5,14). As more comparative analyses are conducted, additional genetic differences will probably be identified between isolates of the two lineages.
In summary, automated sequence analysis of sic and a region of the chromosome with DR sequences permitted rapid and unambiguous differentiation among serotype M1 isolates during a period of a significant increase in the number of invasive disease cases. Genetic analysis of these polymorphic markers permitted us to rapidly rule out the idea that a single unusually virulent strain of M1 GAS was responsible. The subtyping methods described in this work will assist other outbreak investigations and studies designed to understand the molecular basis of temporal variation in disease frequency and severity of infections caused by M1 GAS isolates.
We thank C. Stager, S. Rossman, K. Krause, and C. Baker for generously providing strains.
This work was supported by Public Health Service Grant AI-33119 to J.M.M.
Dr. Hoe is a research associate in the Institute for the Study of Human Bacterial Pathogenesis, Baylor College of Medicine. Her main interests are in the areas of molecular epidemiology and bacterial pathogenesis.
Address for correspondence: James M. Musser, Institute for the Study of Human Bacterial Pathogenesis, Department of Pathology, Baylor College of Medicine, One Baylor Plaza, Houston, Texas 77030, USA; fax: 713-798-4595; e-mail: jmusser@bcm.tmc.edu.
![]()
Top of Page | Current
Issue | Upcoming Issue | Past Issue | Search
| Home