 |
 |
 |
|
 |
FDA Home Page | Search FDA Site | FDA A-Z Index | Contact FDA
PDF Version [767 KB]
U.S. Department of Health and Human
Services
Food and Drug Administration
May 2008
Message from the Commissioner
Executive Summary
Our Approach To Monitoring Product Performance Is
Evolving
The Science of Safety
Information Technology—Key to Modernization
FDA Focus on Safety
Risk Identification
Risk Assessment
Risk Minimization
Laying the Groundwork
The Sentinel Initiative—A National Strategy
for Monitoring Medical Product Safety
Structure/Initial focus
Sentinel Initiative Organizational Challenges
Research and Analytical Capacities
Next Steps
Conclusion
Attachment: Related Federal/Private Sector Activites
![]()
Imagine a national electronic safety system capable of tracking the performance of a drug or medical product, beginning with the earliest stages of clinical research through its effects on millions of Americans who use it to treat or to recover from an illness or condition.
The U.S. Food and Drug Administration of the 21st century needs such an electronic system to serve as sentinel over the safety of medical products and help FDA fulfill its responsibility to protect the health and well being of the American people. Learning all we can about the risks and benefits of medical products is essential. Accurate and reliable information must be obtained before products are approved and afterwards when they are being used by large and diverse populations.
The FDA works hard to learn all we can about the risks and benefits of medical products, beginning before they are approved and continuing after they reach the market. Nevertheless, uncertainties about the safety of medical products regulated by the FDA will always remain. Once a product goes on the market, additional information about the possible risks of its use can almost always be gained. Postmarket safety monitoring is a critical part of our job, and we analyze this information to help guide the best uses of medical products.
What would an electronic system to monitor product safety look like? It would need to have the capability to draw data from sources like electronic health records and medical claims. It should strengthen the ability of the FDA to query other systems quickly and securely for relevant product safety information, within appropriate privacy guidelines. The system also could support research and epidemiology studies and the Agency's existing risk identification and analysis processes. It's possible that such a system could provide a framework for new ways to widely disseminate timely medical product and health-related information.
One of the six key topics identified for action as part of FDA's Critical Path Initiative is harnessing bioinformatics, and FDA has been working to create a wholly electronic environment for the management of its product information, including safety information. In passing the Food and Drug Administration Amendments Act of 2007, Congress has set the stage for making an electronic safety system a reality.
FDA's focus on safety and the promise of collaborating with other experts are critical to creating a successful safety system. The Sentinel Initiative's value to other ongoing medical product performance activities, such as FDA's Critical Path Initiative, makes it an invaluable asset in helping the FDA make the best possible regulatory decisions with the goal of protecting and promoting public health.
This report provides an overview of the projects already under way and outlines the Agency's vision and proposed next steps in the creation of a public-private partnership that could design and implement a national strategy for monitoring medical product safety.
Andrew C. von Eschenbach, M.D.
Commissioner of Food and Drugs
![]()
As we move into the 21st century, we have already begun modernizing our approach to managing health-related information. The U.S. Department of Health and Human Services, under the counsel of the Office of the National Coordinator for Health Information Technology (ONC), has launched a series of initiatives with input from the American Health Information Community.1 These initiatives involve applying information technologies to the way we collect, manage, and share health-related information.2 Technologies such as electronic health records (EHRs), e-prescribing, and electronic decision support tools will make our risk management systems more efficient, improve our ability to protect the public, and, potentially, reduce healthcare costs.
The Food and Drug Administration is playing a key role in this effort. During the past several decades, FDA has taken the lead in developing and implementing standards and terminologies to help manage the regulated product data received as part of the product approval and postmarket surveillance processes. Since the formation of the Healthcare Information Technology Standards Panel, the FDA has coordinated its efforts closely with the standards harmonization process. FDA is also making strides, in part because of the Critical Path Initiative, toward achieving an entirely electronic environment for information management.3 Soon FDA will be able to receive, analyze, and disseminate important health information wholly electronically.
Today, with tools like the Internet, we are able to transmit important health information more quickly and to a wider audience than ever before. As a result, the public is taking increasing interest in health-related matters and assuming more responsibility for their healthcare decisions in a shared process with their doctor or health care professional. A natural and important piece of this equation is consumers' growing interest in the safety of the medical products they use (e.g., medicines, vaccines, devices).
As a nation, we are increasingly focusing healthcare concerns on safety and quality. For example, the Institute of Medicine (IOM) has published a number of reports on how to improve the safety of medical product use. In 1999, IOM issued a landmark report, To Err is Human; a second, related report, Crossing the Quality Chasm, followed in March 2001.The reports describe a vision for improving healthcare quality, patient safety, and the safe use of drugs and medical devices. The reports make clear that a modernized medical product safety system must establish robust links with quality and safety managers and researchers within the broad healthcare system to enable exchange and feedback of information.
This increased focus on safety and quality is, in part, a result of an emerging science of safety, which combines a growing understanding of disease and its origins with new methods of safety signal detection. In addition, personalized medicine is generating information about the unique genetic and biologic features of individuals. All of these advances have implications for the future of healthcare in the United States.
In September 2005, the HHS Secretary asked FDA to expand its current system for monitoring medical product performance. The Secretary asked FDA to explore the possibility of building on the capabilities of multiple data systemsto augment the Agency's data query capability. Such a step would strengthen FDA's ability, ultimately, to monitor the performance of a product throughout its entire life cycle.
The Secretary recommended that FDA explore:
In 2006, the IOM issued a report, entitled The Future of Drug Safety—Promoting and Protecting the Health of the Public.4 Among other suggestions, this IOM report recommended FDA identify ways to access other health-related databases and create a public private partnership to support safety and efficacy studies. The FDA responded to the IOM report in 2007 and is implementing many of its recommendations.5
In early 2007, as a direct response to the Secretary's request, FDA held a two-day workshop with representatives from the federal government, the pharmaceutical industry, the medical device industry, academia, public and private healthcare facilities, healthcare providers, bioinformatics institutions, and the public to explore the feasibility of creating a national electronic system for monitoring medical product safety, based on multiple, broad-based partnerships, including public-private partnerships. The discussion indicated broad support of the concept of developing such a process. There was a clear call for change: supplement the current, mostly passive, system for monitoring postmarket adverse events6 with an active surveillance piece that enables linking to electronic data that can be queried and analyzed in accordance with appropriate security and privacy safeguards.
The Food and Drug Administration Amendments Act of 20077 (FDAAA) calls for active postmarket safety surveillance and analysis. As planned, the Sentinel Initiative will fulfill many of the requirements of FDAAA while fulfilling needs of FDA not contemplated by the law's requirements. FDA is launching the Sentinel Initiative with the ultimate goal of creating and implementing the Sentinel System—a national, integrated, electronic system for monitoring medical product safety.
Specifically, Section 905 of FDAAA calls for the HHS Secretary to develop methods to obtain access to disparate data sources and to establish a postmarket risk identification and analysis system to link and analyze healthcare data from multiple sources. The law sets a goal of access to data from 25 million patients by July 1, 2010, and 100 million patients by July 1, 2012. The law also requires FDA to work closely with partners from public, academic, and private entities.
The budgetary impact of the Sentinel Initiative on FDA during FY 2008 and FY 2009 will largely focus on the time and effort of FDA scientists and researchers in conducting the initial planning and analysis of the initiative, and coordinating input from stakeholders.
The Sentinel Initiative is a long-term effort that must proceed in stages. FDA looks forward to initiating a series of discussions on the scientific and policy issues that must be addressed as the partnership is established. In the meantime, in collaboration with the public and private sectors, FDA will begin to evaluate how to capitalize on and integrate pilot projects that are already underway (see Attachment), many of which directly support the ultimate formation of this new distributed data system.
As an immediate next step, FDA recommends that a public meeting be held with goal of identifying specific short- and long-term plans for establishing the new system. FDA expects to issue a project plan in the upcoming months.
The Food and Drug Administration has the responsibility of regulating medical products—overseeing the development, approval, and postmarket monitoring and surveillance of the drugs, biologics, and medical devices that the American public relies on to maintain and improve their health. FDA must also ensure that information about the performance of a medical product is available to both healthcare professionals and their patients so they can make fully informed choices and use these medical products as safely and effectively as possible.
Using medical products brings benefits and risks. Although marketed medical products are required by federal law to be safe for their intended use, safety does not mean zero risk. A safe product is one that has acceptable risks, given the magnitude of the benefit expected in a specific population and within the context of alternatives available. FDA carefully considers all the available safety information submitted to the Agency during the preapproval process. However, unexpected and sometimes serious safety problems can emerge once a product goes on the market and is used by millions of people.
At present, as part of its safety surveillance and monitoring efforts, FDA relies primarily on (1) health professionals or patients who experience serious problems that they suspect are associated with the drugs and medical devices they prescribe, dispense, or use and submit a report to the Agency or to the manufacturer (who must then report to the Agency); (2) case reports published in the medical literature; and (3) results of postapproval and other clinical studies when they are performed. However, in recent years, rapid scientific advances as well as advances in information technology have created new opportunities for monitoring the performance of medical products.
We are seeing the emergence of a science of safety. This science combines the growing understanding of disease and its origins at the molecular level (including understanding of adverse events resulting from treatment) with new methods of signal detection, data mining, and analysis, enabling researchers to generate hypotheses about, and confirm the existence, and causal factors, of safety problems in the populations using the products. In addition, personalized medicine is generating information about the unique genetic and biologic features of each person that some day will help determine how he or she responds to treatment. Using these tools, FDA has increasingly adopted a life-cycle approach to product development and evaluation. This kind of approach should be used for all medical products so that safety signals generated at any point in the process can be evaluated along with relevant benefit-risk data to inform treatment choices and regulatory decision making. FDA regards improving risk and benefit analysis to be one of the important facets of the science of safety that urgently requires additional development.
The science of safety also offers new opportunities for addressing a fundamental dilemma: the trade off between safety and access. A clear example of this occurs when FDA, after analysis of adverse events, considers whether or not to withdraw a drug from the market for safety reasons. Although withdrawal would eliminate the possibility of further adverse events, it would also deprive those patients for whom the drug is effective of its benefits. If, using methods developed in this new science, we can determine that an adverse event is restricted to a small, identifiable segment of the population, the drug, biologic, or device could remain on the market and continue to benefit those who are not subject to the event.
New collaborations and the application of a systems approach to monitoring postmarket medical product safety are being augmented by the ongoing transformation of the nation's healthcare environment from a paper-based to an increasingly electronic environment. The modernization of the healthcare system is greatly improving our ability to electronically capture and store data, transform it through analysis to new knowledge, then disseminate it to those who need to know. Close to real-time, active surveillance of medical products during routine patient care in a variety of settings may help expand our ability to identify, in a timely fashion, previously unknown risks of medical products, learn about their patterns of use, and assess the outcomes associated with them.8
To achieve a modern healthcare information environment, we need to enhance
and integrate three key information management domains:
(1) access to information; (2) interface, or user-friendly
tools, supported by a robust architecture, to efficiently convert information
into knowledge; and (3) standards. These three domains interact to
influence the way we receive, manage, and communicate information. Improving access to
data sources alone is not enough. We need better interface tools,
and they cannot work efficiently without standards.
Today, much of the safety information collected is largely nonstandardized, making even the most basic analyses and data mining efforts difficult and time-consuming. The lack of a standard format for the exchange of safety data impedes the development of analytic tools and slows the exchange of information among key stakeholders, making it cumbersome and manually labor intensive. In addition, the development and use of terminology standards for important data elements in safety reporting could greatly facilitate data mining and analysis. By developing, adopting, and implementing common data standards, we are establishing a common language for managing and exchanging health-related information, including research and study data. Standards help people communicate successfully. In the information technology world, standards enable electronic information management systems to communicate effectively and efficiently. We must also agree on standard terminology: formal and consistent definitions as well as examples so that we can effectively share concepts and ideas. Standardizing data elements and terminologies is a critical component to any attempt to achieve a modern electronic approach to monitoring medical product performance.
FDA has been proactive during the past decades in the development of medical product terminology and data exchange standards both nationally and internationally. FDA is working closely with the Healthcare Information Technology Standards Panel (HITSP),9 established by the Office of the National Coordinator for Health Information Technology, which seeks to harmonize standards by eliminating unnecessary duplication among health IT standards and by filling gaps between existing standards. HITSP provides input to the American Health Information Community10 and has contributed terminology standards for medication terminology. As part of the Critical Path Initiative, launched in 2004, the Agency made harnessing bioinformatics—including for use in managing the receipt, analysis, and storage of safety information—one of its top Critical Path priorities.
On an international level, FDA has been collaborating with other regulators in promoting standards for human drugs, animal drugs, and medical devices.11 FDA also works closely with both national and international Standards Development Organizations, including Health Level Seven,12 National Council for Prescription Drug Programs, and the International Organization for Standardization. FDA led the development of Structured Product Labeling (SPL), a standard for the exchange of the content of labeling information, and the Individual Case Safety Report (ICSR), a standard for the exchange of adverse event reports, both accredited by the American National Standards Institute. In addition to continuing work in standards development, it is critical that we implement these standards into existing healthcare-related processes across health-related systems. Doing this will require the involvement of relevant health information stakeholders nationwide.
Once data and terminologies have been standardized, we need to ensure that the appropriate tools (interfaces) are available to access the information. We must design and make widely available user-friendly tools for analyzing information that efficiently and effectively convert information into knowledge. We must also make sure users are properly trained to use these tools.
The safety of the medical products regulated by FDA has been a key focus of the Agency since it was established more than a century ago as the nation's first consumer protection agency. Products are approved after FDA determines in the premarket phase that a product's benefits outweigh the risks associated with its labeled use for the intended population. Although FDA has one of the most rigorous preapproval processes in the world, well-conducted, randomized, controlled clinical trials cannot uncover every safety problem, nor are they expected to do so. In most cases, clinical trials aren't large enough, diverse enough, or long enough in duration to provide all the information on a product's performance and safety. In addition, clinical trials are unlikely to reliably detect rare, serious adverse events that occur with long latency or in subpopulations who have not participated in studies. Furthermore, as new medical products enter the market, the potential for interactions with other drugs, biologics, medical devices, and foods increases. Additional information about a medical product almost always can be obtained during postapproval use.
Adverse events can result from a number of causes (when known to be caused by a medical product, they are called adverse reactions). It is estimated that more than 2 million U.S. residents are harmed annually as a result of errors in the prescribing, selection, or use of a prescription or over-the-counter drug (medication error), or because patients experienced a known side effect, problem with the drug's manufacture, or a yet-to-be identified drug-related problem. It is estimated that as many as 100,000 of these episodes result in death annually.13 The rate of adverse events associated with the use of medical devices may be comparable.14 Individuals with multiple health problems are often at greatest risk, particularly older Americans (e.g., Medicare recipients).15
Although manufacturers are required to submit to FDA all reports they receive of adverse events,16 FDA relies to a large extent on the public— both healthcare professionals and their patients—who voluntarily report (to either manufacturers or the FDA) adverse events, errors, and quality problems they observe during the use of a product (so called passive surveillance). Analysis of adverse event reports obtained through passive surveillance is useful for developing hypotheses about possible adverse events that are uncommon and unanticipated, and, in the case of drugs and biologics, for those events that follow soon after the initiation of use or change in dose. Passive surveillance is also the principal mechanism by which signals of rare, but serious, adverse events are currently detected.
With the emergence of the science of safety and the availability of increasingly powerful information technologies, FDA has begun building additional capabilities into its process for monitoring medical product performance. During the past several years, FDA has begun to expand its risk management processes by expanding data source identification and data query capabilities, improving risk identification, assessment, and mitigation strategies, and continuing to work on standards development and implementation efforts.
These activities as well as efforts under way at other agencies and in the private sector will contribute experience directly to the development of the Sentinel System.
In the sections that follow, some key FDA activities under way are discussed within the areas of risk identification, risk assessment, and risk minimization. For a more comprehensive look of relevant federal and private-sector efforts, see the Attachment.
FDA's adverse event reporting program receives information about many medical products and from a variety of voluntary reporters (e.g., prescribers, dispensers, and patients) and has alerted the nation to many important medical product risks. However, the current, mostly passive, system has inherent limitations, notably underreporting of suspected adverse events, incomplete information to enable assessment of causality, and lack of a denominator (i.e., the inability to determine what percent of the population was exposed and what percent experienced the adverse events). To address these limitations, FDA has developed cooperative agreements, contracts, Memoranda of Understanding (MOUs) and other types of collaborations with various members of the public and private sectors. The goal is to strengthen FDA's internal expertise and surveillance data with formalized access to additional data. The short-term focus has been to make available, on relatively short notice, large, population-based databases that will enable rapid access to data in support of studies that address safety issues of concern.
For example, FDA's Medical Product Safety Network (MedSun) is an adverse event reporting program launched in 2002 by FDA's Center for Devices and Radiological Health (CDRH). The primary goal of MedSun is to work collaboratively with the clinical community to identify, understand, and solve problems with the use of medical devices. More than 350 healthcare facilities, primarily hospitals, participate in the entire MedSun network. The program also contains sub-networks including KidNet, an effort to focus on the capture of postmarket safety information from pediatric ICU's and neonatal ICUs; and HeartNet, capturing information from electrophysiology laboratories. LabNet, which captures data from hospital laboratories, has grown to 25 sites. HomeNet (home-use device problem collection) and SightNet (collection of data concerning ophthalmic devices) are in the planning stages.
In another 2007 initiative, FDA signed separate MOUs with the Veterans Health Administration (VHA) and the Department of Defense (DoD). The goals of the VHA collaboration are to explore ways to promote efficient use of tools and expertise for product risk identification, validation, and analysis and to build infrastructure and processes that meet shared needs for evaluating the safety, efficacy, and use of drugs, biologics, and medical devices. The DoD collaboration goals are to explore opportunities to develop, refine, and validate methods for automated safety signal generation using data mining tools with DoD's ALTHA electronic medical record. We are also exploring opportunities to use both data sources for influenza vaccine safety surveillance activities.
Under another FDA–VHA MOU, FDA's Center for Biologics Evaluation and Research is examining methods for using the VHA's VistA electronic health records database to identify influenza vaccine use as well as outcomes that may be vaccine adverse events.
FDA and the National Institutes of Health will soon announce a collaboration to develop the MedWatchPlus Web-based reporting application, an interactive reporting tool to enable healthcare professionals and the public to report suspected adverse experiences associated with any FDA-regulated product.
FDA is working to make existing processes for monitoring medical product performance more robust. Because many of the reports come to FDA voluntarily, they do not facilitate the evaluation of the rate, or the impact, of known adverse events, or the comparative assessment of outcomes for the use of various products. To address these limitations, FDA has sought formalized access to non-Agency epidemiologists and other experts and relevant data sources. For example, FDA's Center for Drug Evaluation and Research currently has contracts with four healthcare system databases, 17 as well as with one U.K. electronic medical records database, to perform secondary analysis to investigate and confirm signals generated through passive surveillance.
As part of the Critical Path Initiative,18 FDA has facilitated the creation of collaborations that are exploring new approaches to analyzing the causes of adverse events. For example, the Severe Adverse Events (SAEs) Consortium, established by the Pharmaceutical Biomedical Research Consortium, is working on developing the patient/sample networks and related research programs required to understand the genetic basis of drug induced serious adverse events. The SAEs Consortium is trying to improve the safety profile of compounds in preclinical and clinical development as well as in drugs already on the market. FDA is acting in a scientific advisory role to the SAEs Consortium.
FDA's Center for Devices and Radiological Health is collaborating with the Agency for Healthcare Research and Quality (AHRQ) to support and provide direction to a multi-center study of the adverse outcomes of late cardiac stent thrombosis. The work is to be conducted within the Developing Evidence to Inform Decisions about Effectiveness (DEcIDE) research network19 and will assess thrombosis risk in relation to various clinical sub-groups and use of antiplatelet therapy. The DEcIDE Network is composed of research centers that AHRQ created in 2005. The network conducts accelerated practical studies about the outcomes, comparative clinical effectiveness, safety, and appropriateness of healthcare items and services. DEcIDE is made up of research-based health organizations with access to electronic health information databases and the capacity to conduct rapid turnaround research. Two AHRQ-funded, pilot DEcIDE projects focus on the establishment, governance, and research use of distributed networks for comparative effectiveness and safety studies of therapeutics and therapeutic interventions. The pilots are in ambulatory, small-practice settings, hospitals, and health plans (see abstracts in Appendix section).
Once an unsuspected risk of harm has been identified, confirmed, and characterized by secondary analyses, FDA has processes in place to help minimize the potential for harm while continuing to facilitate the safe use of a product for those patients for whom it will be beneficial.20
Recently, FDA published a guidance for industry on Risk Minimization Action Plans (RiskMAPs) and held a two-day workshop, co-sponsored with AHRQ, to gain input from stakeholders on how to improve the RiskMAP process and how to use risk minimization tools more effectively.21
Risk minimization tools are intended to minimize known risks. Tools fall in one of three categories.
Tools in this category employ specific, targeted education and outreach efforts about risks to increase appropriate knowledge and behaviors of key people or groups (e.g., healthcare practitioners and consumers) that have the capacity to prevent or minimize the product risks of concern.
These tools can be used in addition to efforts in the targeted education and outreach category. Examples include systems that prompt, remind, double-check, or otherwise guide healthcare practitioners and/or patients in prescribing, dispensing, receiving, or using a product in ways that minimize risk.
Performance-linked access tools include systems that link product access to laboratory testing results or other documentation.
In 2006, FDA's Center for Devices and Radiological Health issued a report on Ensuring the Safety of Marketed Medical Devices: CDRH's Medical Device Postmarket Safety Program. This report outlines an initiative that brings all of the needed center experts together to consistently monitor information about the use of medical devices and to expeditiously address risk mitigation approaches for devices. CDRH is organizing matrix networks with expertise in specific categories of medical devices. The networks will suggest new methods for identifying, analyzing, and mitigating risks associated with the use of medical devices. Information about these risks and the mitigation strategies will be shared with manufacturers for use in the development of the next generation of medical devices.
FDA also has at hand a number of new risk communication tools, using a variety of electronic communication channels, to provide timely safety information to both the healthcare community and patients. Physicians, pharmacists, nurses, and patients and other consumers can now receive safety alerts directly from FDA by e-mail listserve notification, RSS feeds,22 audiopodcasting, or streaming video.23 FDA's MedWatch program maintains a Partners program24 with more than 100 professional organizations who work with the Agency to support the voluntary reporting of adverse events and the dissemination of timely safety information to their members.
In June 2006, FDA implemented new labeling requirements for the content and format of FDA-approved prescribing information (package inserts). The new design helps manage the risks associated with prescription drug use by providing the prescriber with the most up-to-date information in an easy-to-read format. The format includes a “Highlights” section that summarizes the most critical prescribing information and a table of contents that refers prescribers to detailed information located in the labeling. In addition, the content of the new package insert includes a list of recent changes made to the labeling, including new warnings, a toll-free telephone number for adverse event reporting, and a separate section that contains information for counseling patients about a drug's risks.
All of these risk identification, analysis, and minimization efforts, as well as the partial listing of activities in the Attachment, support the Sentinel System concept and could, ultimately, be integrated into the system.
In 2004, the President established the Office of the National Coordinator to provide counsel to the HHS Secretary and departmental leadership for the development and nationwide implementation of an interoperable health information technology infrastructure.25 In 2005, the American Health Information Community, a federal advisory committee made up of public and private sector leaders who represent a broad spectrum of healthcare stakeholders, was chartered to make recommendations to the HHS Secretary on how to accelerate the development and adoption of health IT, make health records digital and interoperable, encourage market-led adoption, and ensure that the privacy and security of those records are protected at all times.
In September 2005, the HHS Secretary directed FDA to expand its current system for monitoring medical product performance by capitalizing on the emerging sciences of information technology and safety and exploring the potential for adding an active surveillance capability to current passive surveillance systems. In March 2006, FDA identified harnessing bioinformatics as one of its top six Critical Path priorities.26 After meetings with stakeholders, in March 2007, FDA held a two-day public workshop on the idea of creating a nationwide system for monitoring medical product safety, a sentinel system.27 The idea of creating a nationwide system was met with overwhelming support. Such a system would consist of an architecture that would enable FDA (and ultimately other researchers) to query remote data sources (maintained by their owners), using appropriate security and privacy safeguards, for specific medical product information. Participants noted that a number of key issues would have to be resolved:
With the passage of the Food and Drug Administration Amendments Act of 2007,28 FDA now has the mandate to launch this initiative. Section 905 of the FDA Amendments Act explicitly directs the Secretary (in collaboration with public, academic, and private entities) to develop methods to obtain access to disparate data sources and to validate methods for the establishment of a “postmarket risk identification and analysis system” to link and analyze safety data from multiple sources.29
Section 905 sets a goal of accessing data from 25 million patients by July 2010, and 100 million patients by July 2012. To achieve these goals, FDA and others will need to invest in the planning, design, and implementation of systems and interfaces.
Because of the significant challenges inherent in developing such a system (e.g., risk of generating false positive signals, potential of misperception of such signals), FDA will take a staged approach to the development of this new system, using funds within the FY 2008 appropriation. Funding for these activities for FY 2009 are included in the President's requested budget. In the meantime, relevant activities under way at FDA, some as part of the Critical Path Initiative launched in 2004,30 will continue, as will related efforts initiated independently by other agencies or by the private sector. A number of these activities, which FDA is participating in or observing, can serve as proof-of-concept studies and pilots.
As described in the previous section, many projects already are under way at FDA and in the private sector that take advantage of available data sources to identify and analyze signals that may indicate medical product safety problems. This experience and the many existing cooperative activities that have been launched during the past decade reflect our increasing ability to share and compare data from other sources—we are also seeing how valuable such a collaborative approach is.
Consequently, in response to the recommendations from the IOM, to the request from the HHS Secretary, to provisions included in the recently enacted FDA Amendments Act, and in close coordination with the Office of the National Coordinator for Health Information Technology, FDA is launching the Sentinel Initiative. The goal of the Initiative is to create a national, integrated, electronic system (the Sentinel System) for monitoring medical product safety. The Sentinel System, which will be developed and implemented in stages will ultimately enable us to access the capabilities of multiple, existing data systems (e.g., electronic health record systems, medical claims databases)to augment the Agency's current capability. The System will enable us to query distributed data sources31 quickly and securely for relevant de-identified product safety information. This will strengthen FDA's ability to monitor the performance of a product, eventually, throughout its entire life cycle. Such a system could also ultimately facilitate data mining and other research-related activities.32 The Sentinel System will facilitate targeted queries, within the bounds of established privacy and security safeguards, across remote systems and be scalable to enable small or large queries using broad or narrowly focused data. The Sentinel System can be achieved with minimal transfer of data (data sources will be maintained and managed by their owners), using tools and processes that will ensure the protection of personal and proprietary information.
The Sentinel System could also be used in the future to support research on use and outcomes and epidemiology studies as well as existing risk identification and analysis processes. The Sentinel System could ultimately even support the development of new pathways for dissemination of up-to-date medical product and health-related information.
The Sentinel System will build on existing systems and data, to the extent
practicable, rather than create a new system; follow scientific principles
of surveillance; use health IT standards harmonized by HITSP considered by
the American Health Information Community and recognized by the Secretary;
and ensure the protection of privacy and security of
personal health information. Part of the Initiative will be to explore use
of
the emerging Nationwide Health Information Network which, once developed, will
have the ultimate goal of interconnecting clinicians across the healthcare
system and enabling the sharing of data as necessary with public health agencies.
Nationwide Health Information Network (NHIN) The NHIN is the portion of the health IT agenda intended to provide a secure, nationwide, interoperable health information infrastructure that will connect providers, consumers, and others involved in supporting health and healthcare. The NHIN will enable health information to follow the consumer, be available for clinical decision making, and support appropriate use of healthcare information beyond direct patient care so as to improve health. The NHIN will be a built out of state and regional health information exchanges (HIEs) and other networks so as to support the exchange of health information by connecting these networks and the systems they, in turn, connect. The NHIN seeks to achieve these goals by:
|
We propose launching the Sentinel Initiative through a broad-based public-private partnership, building on HHS investments in health information technology programs that have already been launched (e.g., NHIN33 trial implementations; efforts under way as part of the Federal Health Architecture34; standards development and standards recognition efforts; MedWatch; and electronic clinical trial data collection), as well as the experience gained by private and government healthcare systems, academic organizations, and FDA in analyzing data from healthcare databases.
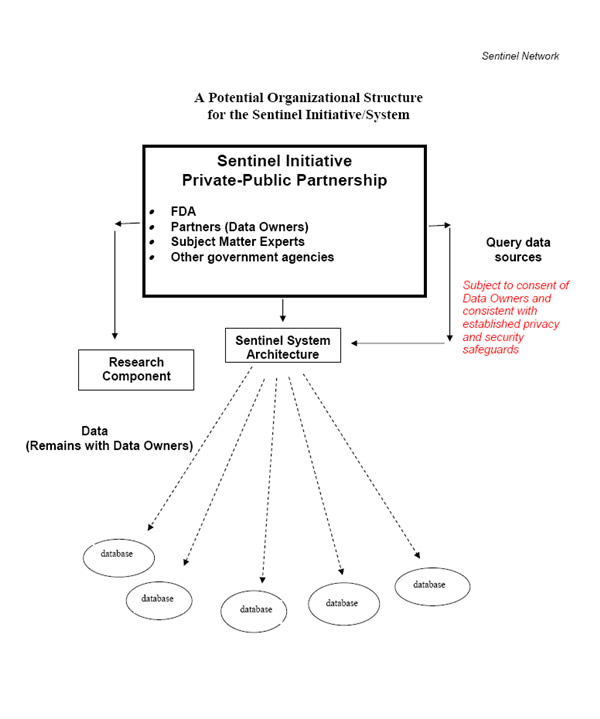
The broad organizational structure and function of the Sentinel Initiative ultimately will consist of several linked components, including:
It is expected that many of the data owners (e.g., healthcare systems, private entities) will be members of the partnership. However, the data owners may choose to provide data under contracts while not belonging to the partnership.
Examples of key activities include:
It will take time to establish the partnership and to set timetables for short- and long-term goals.
Establishment of the Sentinel Initiative and the Sentinel System architecture raises a number of administrative, organizational, procedural, and methodological challenges. We propose the following key principles for the organizational framework.
Privacy Protection and Data Security: Safeguards for protecting data and personal privacy information must be established before the Sentinel System becomes operational.
Scientific Credibility: Scientific analysis must adhere to high standards for data integrity and human subject protection and potential conflicts of interest should be avoided.
Integrity: The management structure and data analysis components of the System must be insulated from undue influence.
Systems Approach: Effective, life-cycle, safety surveillance of medical products requires a systems approach. Private stakeholders in collaboration include regulated industry, healthcare provider communities, and academics in the pharmacovigilance, information management, and risk communication disciplines. Public stakeholders include governmental public health and regulatory agencies, and the public and its elected and appointed representatives. All stakeholders have a specific role to play.
Governance: The governance structure and process should provide for incorporation of a broad range of expertise and opinion and minimize both conflicts of interest and perceptions of conflicts of interest.
Inclusiveness: Stakeholders should have an opportunity to provide input on the standards and processes used by the system.
Transparency: Protocols, data, and study results should be made available to the public.
Open Source: The standards, methodologies, and source codes for software used by the system should be available to end users.
A key activity of the Sentinel Initiative will be to provide for the development of electronic interfaces that can send queries to existing data sources consistent with appropriate privacy guidelines and applicable laws. The ongoing development and deployment of healthcare system-based electronic records for clinical encounters, laboratory, and other diagnostic data occurring both in hospital and outpatient settings offer important opportunities to query a variety of sources quickly.
Improving the interoperability of data queries and data analysis will make these activities more efficient. The system that is developed will enable the sharing of analytical resources, reduce duplicative efforts, and maximize the accuracy of the queries. Efforts already under way elsewhere in the private and the public sectors (e.g., NHIN (described above), also see Attachment for additional examples) and the standards efforts that have been advanced and supported by the HHS Secretary should facilitate the interoperability necessary to effectively perform the medical product surveillance functions of the Sentinel System.
Data mining and other tools and processes, which must be developed, validated, and implemented, will, ultimately help identify or further confirm new potential risks and help epidemiologists establish large study populations for formally testing their safety hypotheses through observational methods.35 Use of these types of tools, in conjunction with population exposure data and other input, could result in more directed application of in-depth epidemiological risk analyses. Standardized data sets and interoperability among databases will facilitate these analyses.
The Sentinel Initiative, which will be a long-term effort, must proceed in stages. FDA will conduct these activities with funding within the FY 2008 appropriation and the FY 2009 President's budget request. FDA looks forward to rapidly initiating a series of discussions on the scientific and policy issues that must be addressed. In the meantime, in collaboration with the public and private sectors, FDA will begin to evaluate how to capitalize on and integrate pilot projects already under way (see Attachment). Most of those projects directly support the ultimate formation of this new distributed data system.
FDA recommends that a public meeting be held in collaboration with potential partners as a next step. The goal should be to formalize specific short- and long-term plans for establishing the new system. FDA would expect to issue a project plan in the coming months.
The time is right for improving the way we generate evidence about medical product performance. With the passage of the Food and Drug Administration Amendments Act, Congress has mandated creation of a medical product safety monitoring system. Many activities already under way, some as part of the Critical Path Initiative, will contribute directly to this broad effort.
The emergence of a new science of safety, strides made in developing and implementing
electronic standards, new systems for managing health information, and FDA's
steady focus on safety and collaboration are keys to creating a successful
Sentinel System for monitoring medical product performance.
The potential for the Sentinel System ultimately to support many other activities
critical to a modern healthcare system is noteworthy. For example, the research
and analytical capabilities possible under the Sentinel Initiative could enhance
the health communication tools FDA currently uses while supporting the creation
of new tools to reach new audiences in new ways. Health researchers may be
able to use the query system created for the Sentinel System to evaluate the
outcomes and quality of various treatments.
The creation of a Sentinel System is a long-term undertaking that will require the commitment of federal and private sector financial and human resources during the coming years. Congress has set rigorous goals, but they are achievable with a concerted effort.
While the broad Sentinel Initiative is being planned and implemented, FDA will continue participating in ongoing pilot projects and, as appropriate, initiate new projects with relevant stakeholders. We will continue working nationally and internationally on standards development efforts. And we will continue ongoing efforts to enhance existing risk management and risk communication processes at FDA. All of these efforts will proceed consistent with the ultimate goals of the Sentinel Initiative.
FDA also eagerly anticipates the discussions on the scientific and policy issues that must be addressed as plans for the Sentinel Initiative move forward.
This is a partial listing of the many federal and private sector activities36 under way that are consistent with the goals of the Sentinel System concept. Activities with FDA involvement are organized into three groups: Risk Identification, Risk Assessment, and Risk Mitigation. Some private-sector activities that were described at FDA's two-day workshop in 2007 are listed following the FDA activities.
Risk Identification
Risk Assessment
Risk Mitigation
Examples of Private Sector Safety Surveillance Efforts (from the FDA two-day Sentinel workshop)
Over the past three decades, Regenstrief research scientists have developed the Regenstrief Medical Records System (RMRS). More than 32 million physician orders have been entered into the computerized order entry system of the RMRS that provides unique clinical decision support and guidelines. RMRS has a database of 3 million patients, with 900 million discrete, on-line results, 16 million text reports including diagnostic studies, procedure results, operative notes, discharge summaries, and 50 million radiology images. The RMRS serves as the day-to-day electronic medical records system at a 319-bed public hospital and its community clinics.
In 1994, with funding from the National
Institutes of Health and the National
Library of Medicine, Regenstrief
Institute Medical Informatics extended the RMRS to
the Indiana Network for Patient Care (INPC), a city-wide clinical informatics
network. INPC covers 15 hospitals in central Indiana and allows physicians
caring for the patient to view as a single virtual record much of a patient's
previous care.
All INPC participants now deliver registration records, all laboratory tests,
and all UB92 records (diagnosis, length of stay, and procedure codes) for hospital
admissions and emergency room visits to separate electronic medical record
vaults in a central INPC server. The computer system standardizes all clinical
data as it arrives at the INPC vault, laboratory test results are mapped to
a set of common test codes with standard units of measure, and patients with
multiple medical record numbers are linked. Each institution has the same file
structure and shares the same term dictionary, which contains the codes, names
(and other attributes) for tests, drugs, coded answers, etc.
Building upon RMRS and INPC, the Indiana Health Information Exchange, Inc., a nonprofit corporation for sharing clinical information among healthcare providers and other health care entities is dedicated to improving the quality, safety and efficiency of healthcare in the state of Indiana.
i3 Aperio is a drug experience registry that marshalls i3 Drug Safety's epidemiological expertise and a proprietary claims database containing the healthcare experiences of more than 11 million individuals—from claim databases maintained by United Health Group (UHG)—to enable users to monitor the safety of new drugs quickly and effectively. Instead of associating disparate databases, i3 Aperio uses an integrated database of pharmacy, hospital, and physician claims information.
By accelerating the acquisition of real-world information on the safety of new prescription drugs following their launch, i3 Aperio can help researchers identify signals that could indicate a drug's potential safety issues at a speed closer to real-time than previously available. i3 Aperio can also help researchers track and assess the potential risks of prescription drugs that have already been on the market for several years.
Anceta, the American Medical Group Association (AMGA), and participating AMGA multispecialty medical groups have been engaged in the development of a national Collaborative Data Warehouse comprising comprehensive, longitudinal patient healthcare information. The Collaborative Data Warehouse will provide the participating medical groups with access to comparative data among medical groups of similar size and structure and key benchmarks for practice management, clinical performance, product performance, health outcomes, economics, and quality of care. Additionally, the Warehouse and its proprietary health informatics tools will assist providers, policy makers and purchasers (including employers and employees) in making better decisions regarding healthcare choices. Anceta is accessing electronic health information from medical groups that have information technology infrastructures currently more advanced than those typically found throughout the nation's general healthcare system. Since these multispecialty group practices “own” a greater portion of a patient's complete healthcare picture, usually with common patient identifiers among ambulatory care and hospital information systems, data can be more easily collected and integrated for data warehousing.
The electronic Primary Care Research Network (ePCRN) is an electronic infrastructure that facilitates the conduct of randomized controlled trials in primary care and promotes the translation of research findings into practice. The electronic infrastructure of the ePCRN is being built on a web-enabled distributed database technology that makes use of cutting-edge web technologies. This allows creation of distributed clinical information systems located at the site of practice that can be appropriately and securely linked together. It provides a highly secure, Internet-based electronic infrastructure that will enable primary care practices anywhere in the United States to link with researchers in academic centers or the National Institutes of Health (NIH) to facilitate recruitment, entry, and follow-up of participants in multidisciplinary trials. The overall goals of the ePCRN are to provide the ability to perform large national collaborative studies throughout the United States, improve efficiency and reduce costs for individual trials, provide easier access for data retrieval and analysis, and involve primary care practices in the discovery and the translation of research findings into practice.
The National Cancer Institute's Cancer Research Network comprises 14 healthcare systems across the nation, all members of the HMO Research Network, which is a larger consortium of health maintenance organizations with formal recognized research capabilities and a shared commitment to public domain research. The Virtual Data Warehouse Project has been developed to increase efficiency by making it possible for programs written at one HMO research site to be run at other HMO sites. This has encouraged consistency and standardization and enabled quicker turnaround of queries.
Examples of Distributed Research Networks: AHRQ DeCIDE-funded projects
To support AHRQ's Effective Health Care program, the DEcIDE centers at the HMO Research Network Center for Education and Research on Therapeutics (HMORN CERT) and the University of Pennsylvania will develop a design and specifications for a scalable distributed research network to support a wide array of purposes related to therapeutics, including comparative effectiveness, safety, and use, as well as quality of care research. They will implement a prototype, conduct a proof of principle research project on hypertension therapy, and make recommendations for future expansion of the network.
The development of the network prototype and implementation of the proof of principle research project will take place in six HMO Research Network health plans: Geisinger Health System, Group Health Cooperative, Harvard Pilgrim Health Care/Atrius Health, HealthPartners, Kaiser Permanente Colorado, and Kaiser Permanente Northern California. The research project will merge data from both electronic medical records and administrative claims data.
This work is intended to inform the development of a larger multi-purpose distributed research network that will include both private and public partners. To further this goal, the investigators will consult and coordinate with national leaders in the development and use of electronic administrative and medical record data systems.
The Distributed Network for Ambulatory Research in Therapeutics (DARTNet) will be a prototype federated network of electronic health record (EHR) data from eight organizations representing more than 200 clinicians and more than 350,000 patients. The prototype system will capture, codify, and standardize a subset of unique data elements per patient for up to 24 months. Project aims are as follows.
Aim 1: Develop a federated network of
more than 200 EHR enabled primary care clinicians and examine network governance,
data extraction, and software implementation.
Aim 2: Demonstrate how existing data sets can be enhanced
by patient-level data from DARTNet clinicians and patients to inform and
expand knowledge of effective and safe medical therapeutics.
Aim 3: Demonstrate the ability to collect specific
data from clinicians, staff or patients on a clinically defined set of individuals
to enrich the EHR data set and answer effectiveness and safety questions
concerning medical therapeutics.
The project will be completed over 15 months and will involve finalizing organizational involvement, refining, and testing the federated data system, identifying data elements, obtaining IRB approval, completing the data mapping process, testing the distributed query processes, developing the governance structure, completing the informatics components, adding DARTNet derived data to the pilot project, completing a research project, and writing reports and manuscripts.
In the research phase of the project, the investigators will use the DARTNet prototype to examine the efficacy and safety of oral diabetes medications for the treatment of type 2 diabetes.
DARTNet will examine the research areas using claims data and will then repeat
the analyses using the DARTNet dataset, which should provide an expanded set
of data elements, but a smaller sample size.
![]()
1The American Health Information Community is a federal advisory body, chartered in 2005, to make recommendations to the HHS Secretary on how to accelerate the development and adoption of health information technology (HIT) and to help advance efforts to achieve the President’s goal for most Americans to have access to secure electronic health records by 2014.
2 See http://www.hhs.gov/healthit/.
3 One of the six key topics of focus for the Critical Path Initiative is the harnessing of bioinformatics to manage regulated product information.
4 Institute of Medicine, The Future of Drug Safety —Promoting and Protecting the Health of the Public, September 22, 2006, http://www.iom.edu/.
5 FDA, The Future of Drug Safety – Promoting and Protecting the Public Health, FDA's Response to the Institute of Medicine's 2006 Report, January 2007, http://www.fda.gov/oc/reports/iom013007.pdf.
6 Any untoward medical event associated with the use of a drug, device, or biologic in humans, whether or not considered product related. When known to be caused by a medical product, these events are called adverse reactions.
7 Food and Drug Administration Amendments Act of 2007, Public Law 110-85, was signed into law in September 2007. See Title IX, Section 905.
8 See, for example, Wadman, M., Nature 446, 358-359 (22 March 2007); Published online 21 March 2007.
9 For background, see http://www.hitsp.org.
10 AHIC is a federal advisory body, chartered in 2005, to make recommendations to the HHS Secretary on how to accelerate the development and adoption of health information technology (HIT) and to help advance efforts to achieve the President’s goal for most Americans to have access to secure electronic health records by 2014.
11These efforts take place under the auspices of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), the Veterinary International Conference on Harmonisation (VICH), and the Global Harmonization Task Force (GHTF), respectively.
12 For example, FDA is working with Pfizer/CDISC over the next year on multi-phase pilot testing for the HL7 ICSR. CDISC has developed a tool that converts source data from a clinical information system to a format that can be transmitted to regulatory authorities to report clinical trial adverse events. The pilot will test this tool using the HL7 ICSR. The testing also involves creating HL7 ICSRs directly from electronic health record systems, and we will be working with Brigham and Women's Hospital to test reporting for postmarket drug and device adverse events.
13 Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998;279(15):1200-5.
14 FDA, Ensuring the Safety of Marketed Medical Devices: CDRH's Medical Device Postmarket Safety Program; January 18, 2006. Available at http://www.fda.gov/cdrh/postmarket/mdpi-report.html.
15 Gurwitz, JH, et al. Incidence and preventability of adverse drug events among older persons in the ambulatory setting JAMA 2003;289:1107-1116.
16 In addition, the MDR (Medical Device Reporting) system, which is transitioning to an electronic version of mandatory report (e-MDR), enables the Agency to view collective reports of adverse events for analysis. Similarly, MedSun is a sentinel-based system that collects real-time information about the use of medical devices and information about adverse events through a network of hospitals engaged in formal agreements with the Agency.
17 Ingenix Inc (UnitedHealth Group); The Vanderbilt University; Kaiser Foundation Research Institute; and Harvard Pilgrim Health Care Inc (HPHC) (HMO Research Network Center for Education and Research on Therapeutics).
18 For more on the Critical Path Initiative and related activities, see http://www.fda.gov/oc/initiatives/criticalpath/.
19 See http://effectivehealthcare.ahrq.gov/aboutUs/generate.cfm.
20 See http://www.fda.gov/cder/guidance/6358fnl.pdf.
21 These processes also are addressed by the FDA Amendments Act provisions regarding risk evaluation and mitigation strategies (REMS), which will replace existing RiskMAPs.
22 RSS (Really Simple Syndication) is a format for electronically syndicating news and the content of news-like sites, including major news sites, personal Weblogs, or anything that can be broken down into discrete items.
23 FDA's “Patient Safety News.”
24 Many private healthcare-related organizations (e.g., professional societies, consumer groups and healthcare media organizations) are partnering with FDA's MedWatch program (http://www.fda.gov/medwatch/Partner.htm) to further disseminate timely safety information to their members and subscribers.
25 The goal of this infrastructure is to improve the quality, safety, and efficiency of healthcare and the ability of consumers to manage their health information and healthcare. For more, see http://www.hhs.gov/healthit/onc/mission/.
26 For more on the Critical Path Initiative, see http://www.fda.gov/oc/initiatives/criticalpath/.
27 FDA, Sentinel Network to Promote Medical Product Safety: Public Meeting, Federal Register, Vol. 72, No. 11, January 18, 2007, Docket No 2007N-0016.
28 Public Law 110-85.
29 The Amendments Act of 2007 requires FDA to (in collaboration with public, academic, and private entities) develop methods to obtain access to disparate sources of data and validated methods to link and analyze safety data from multiple sources (section 905(a) (3) (B) (i) – (ii) (II)).
30 See http://www.fda.gov/oc/initiatives/criticalpath/.
31 Data sources will continue to be owned and maintained by their owners.
32 The Sentinel System could support other efforts, such as on-going efforts to expand evidence-based medicine.
33 For background, see http://www.hhs.gov/healthit/healthnetwork/trial/.
34 Federal Health Architecture was established to create a consistent federal framework to facilitate communication and collaboration among all healthcare entities to improve citizen access to health-related information and high-quality services, part of the President's plan to expand electronic government.
35Other possible activities could included data mining for signal detection, characterization of therapeutic use patterns, and establishing background rates of disease.
36 The appearance of a particular activity in this list does not imply FDA endorsement.
37 For background on NEISS, see http://www.cpsc.gov/library/neiss.html.
38 Ingenix Inc. (UnitedHealth Group); The Vanderbilt University; Kaiser Foundation Research Institute; and Harvard Pilgrim Health Care Inc (HPHC) (HMO Research Network Center for Education and Research on Therapeutics).
39 For more on this agency, visit the AHRQ Web site at http://www.ahrq.gov/.
40 The CERTS program was develop as a result of specific direction provided by Congress in the 1997 Food and Drug Administration Modernization Act.
41 See http://www.fda.gov/bbs/topics/NEWS/2006/NEW01467.html; www.cardiac-safety.org.
42 See http://www.fda.gov/cder/dsn/default.htm.
43 See http://www.fda.gov/cder/guidance/index.htm.
44 Many private healthcare-related organizations (e.g., professional societies, consumer groups and healthcare media organizations) are partnering with FDA's MedWatch program (http://www.fda.gov/medwatch/Partner.htm) to further disseminate timely safety information to their members and subscribers.
![]()