|
|
Background Influenza A (H5N1) viruses could cause a severe worldwide epidemic, with high attack rates, large numbers of deaths and hospitalizations, and wide disruption. Effective vaccines against these viruses in humans are urgently needed.
Methods We conducted a multicenter, double-blind two-stage study involving 451 healthy adults 18 to 64 years of age who were randomly assigned in a 2:2:2:2:1 ratio to receive two intramuscular doses of a subvirion influenza A (H5N1) vaccine of 90, 45, 15, or 7.5 µg of hemagglutinin antigen or placebo. The subjects were followed for the safety analysis for 56 days. Serum samples obtained before each vaccination and again 28 days after the second vaccination were tested for H5 antibody by microneutralization and hemagglutination inhibition.
Results Mild pain at the injection site was the most common adverse event for all doses of vaccine. The frequency of a serum antibody response was highest among subjects receiving doses of 45 µg or 90 µg. Among those who received two doses of 90 µg, neutralization antibody titers reached 1:40 or greater in 54 percent, and hemagglutination-inhibition titers reached 1:40 or greater in 58 percent. Neutralization titers of 1:40 or greater were seen in 43 percent, 22 percent, and 9 percent of the subjects receiving two doses of 45, 15, and 7.5 µg, respectively. No responses were seen in placebo recipients.
Conclusions A two-dose regimen of 90 µg of subvirion influenza A (H5N1) vaccine does not cause severe side effects and, in the majority of recipients, generates neutralizing antibody responses typically associated with protection against influenza. A conventional subvirion H5 influenza vaccine may be effective in preventing influenza A (H5N1) disease in humans. (ClinicalTrials.gov number, NCT00115986
[ClinicalTrials.gov]
.)
These viruses possess a new H5 subtype of hemagglutinin, against which at present there is little immunity in human populations. The viruses have the potential to cause extremely severe respiratory illness in humans, and of the 169 cases reported to the World Health Organization as of February 13, 2006, 91 (54 percent) have been fatal.3 Many of the viruses isolated from humans have been found to be genotypically resistant to the adamantanes,4 and resistance to oseltamivir has also been described.5 Although human-to-human transmission appears at present to be rare,6 the development of an effective vaccine against influenza A (H5N1) virus is a matter of considerable urgency.
Inactivated influenza vaccines used annually for the control of human influenza are made from purified virions grown in embryonated hens' eggs and are formulated to contain not less than 15 µg of the hemagglutinin of the represented strains per 0.5-ml dose; the vaccine is administered intramuscularly without adjuvant. Efficacy in adults for the prevention of laboratory-confirmed influenza is typically in the range of 70 to 90 percent.7,8 An influenza A (H5N1) vaccine that contained a new antigen but was otherwise similar to licensed vaccine could be considered by regulatory authorities to represent a change in strain, rather than a completely new product, thereby facilitating the rapid licensure of the vaccine. Therefore, we evaluated an egg-grown, inactivated subvirion H5 vaccine administered without adjuvant. Because an immunologically naive population would probably require a two-dose schedule of administration, as is used in children, and might require higher doses than are used in annual influenza vaccinations, we evaluated a two-dose regimen, with doses ranging from 7.5 to 90 µg.
Methods
Vaccine
The seed virus for the production of the experimental influenza A (H5N1) vaccine was generated from the human isolate influenza A/Vietnam/1203/2004 (H5N1) virus with the use of a plasmid rescue system.9 The gene segments encoding the hemagglutinin and neuraminidase were derived from the A/Vietnam/2004 virus, and all other genes were derived from the A/PR/8/34 virus, a laboratory strain commonly used as a platform for influenza vaccines. The hemagglutinin gene was further modified to replace the stretch of six basic amino acids at the cleavage site between hemagglutinin 1 and hemagglutinin 2, associated with high pathogenicity in birds with an avirulent avian sequence. The resulting influenza rgA/Vietnam/1203/2004xA/PR/8/34 influenza (H5N1) virus was antigenically identical to the wild-type A/Vietnam/1203/2004 virus, reached high titers in eggs, and was apathogenic in chickens, allowing the virus to be handled under the containment conditions of a biosafety level 2 laboratory, with additional safety precautions.
The vaccine product was generated according to standard techniques. The seed virus was grown to a high titer in eggs, the virions were purified by means of centrifugation, inactivated with the use of formalin, disrupted with the use of Triton X-100, and filtered to remove bacteria. The vaccine underwent further purification and was formulated at concentrations of 90 µg of hemagglutinin protein per milliliter (lot U10915C) and 30 µg of hemagglutinin protein per milliliter (lot U10914C) in vials containing 0.7 ml without preservative. The content of immunologically active hemagglutinin in the final formulation was determined with the use of single-radial-immunodiffusion. Sheep antiserum to bromelain-cleaved native H5 hemagglutinin was used in the agar. Placebo consisted of normal saline. Both vaccine and placebo were stored at 4°C until use.
Study Design
We conducted a randomized, placebo-controlled, double-blind, multicenter trial. Written informed consent was obtained from potential subjects. Healthy adults 18 to 64 years of age were carefully screened for the absence of any chronic illnesses (enrollment criteria are described in detail in the Supplementary Appendix, available with the full text of this article at www.nejm.org). Eligible subjects were randomly assigned in a 2:2:2:2:1 ratio to receive two doses of vaccine at a dose of 90, 45, 15, or 7.5 µg or placebo. Each dose was administered intramuscularly into the deltoid muscle, and the two doses were given 28 days apart. A permuted-block randomization was used, and subjects were stratified according to study site, age (18 to 39 years vs. 40 to 64 years), and history of receipt of the 2004 formulation of licensed influenza vaccine. All vaccinations were administered by a clinician who was not involved in the assessment of adverse events or the laboratory follow-up, and the contents of the syringe were shielded from the subject's view. The subjects were observed for 30 minutes after the receipt of each dose for adverse events, and for the next seven days, they recorded the presence and severity of local symptoms (pain and tenderness) and systemic symptoms (feverishness, malaise, myalgia, headache, and nausea) and oral temperature. Subjects used a standard scale to grade side effects during this seven-day period, in which symptoms were considered mild if they did not interfere with normal activities, moderate if they resulted in some interference with normal activities, and severe if they prevented subjects from engaging in normal daily activities.
The subjects' observations were reviewed by members of the study staff on day 7 after each vaccination, and the medical history and record of adverse events occurring during the interval were also reviewed on days 28 and 56. Samples of serum for the assessment of antibody responses were obtained before each vaccination and again 28 days after the second vaccination.
The study was conducted in two stages. In the first stage, 118 subjects were enrolled and followed, as described. In addition, hemoglobin levels, total white-cell count, platelet count, creatinine levels, and serum alanine aminotransferase levels were determined in each subject before vaccination and on day 7 after each vaccination. After the safety data through the seven days after the first dose were reviewed by an independent data and safety monitoring board, the remaining 333 subjects were enrolled and treated, as described, but without the additional laboratory measurements. A second review of the initial group of 118 subjects was performed before the second dose was administered to the remaining subjects.
Laboratory Assays
Microneutralization assays and hemagglutination-inhibition assays were performed at a central laboratory (Southern Research Institute) with the use of the influenza rgA/Vietnam/1203/2004xA/PR/8/34 influenza (H5N1) vaccine. In addition, a subgroup of samples were also tested with the use of the wild-type influenza A/Vietnam/1203/2004 virus under conditions of enhanced biocontainment (biosafety level 3-plus laboratory). Microneutralization assays were performed as described previously.10,11 Serum samples were tested at an initial dilution of 1:20, and those that were negative were assigned a titer of 10. Serum samples were tested separately and in duplicate; if the results showed a difference by a factor of 2, the samples were retested.
Hemagglutination-inhibition assays were performed according to established procedures,12,13 with the use of horse erythrocytes. After treatment with receptor-destroying enzyme to remove nonspecific inhibitors of agglutination, the serum samples were tested at an initial dilution of 1:20.
Statistical Analysis
The prespecified primary immunologic end point of the trial was the proportion of subjects in each group categorized according to dose of vaccine or receipt of placebo in whom a neutralizing titer of 1:40 or greater developed against the rgA/Vietnam/1203/2004 virus at day 28 after the administration of the second dose of vaccine. The geometric mean of duplicate results for each specified time was used for the calculation. Exact (Clopper–Pearson) confidence intervals are reported for all proportional end points. Geometric mean titers of antibody and their confidence intervals were computed by transforming the results to a logarithmic scale, assuming asymptotic normality conditions were satisfied on the scale and converting back to the original scale.
Rates of reactogenicity after each vaccination were based on the most severe response reported. The rates were compared by an exact linear-by-linear–association test (with the use of antigen dose levels and evenly spaced scores for reactogenicity). The overall comparison between vaccine and placebo for reactogenicity was based on an exact permutation test in which reactogenicity was dichotomized as none to mild or moderate to severe. The antibody dose–response relationship was assessed with the use of a general linear model. Comparisons of geometric mean titer between groups were performed with the use of the Wilcoxon rank-sum test, and response rates were compared with the use of the Mantel–Haenszel chi-square test. Spearman's correlation coefficient was used to assess the correlation in neutralization titers to different viruses. All reported P values are two-sided. StatXact software, version 6.1 (Statistical Solutions), was used to compute the exact tests. All other data manipulations and statistical computations were conducted with SAS software, version 8.2.
The sample size for this study (100 subjects in each vaccine group and 50 subjects in the control [saline] group) was selected to provide a robust initial safety database as well as some information on the dose-related immune response in a timely fashion. Given the enrollment of 100 subjects in each vaccine group, the half-width of a 95 percent confidence interval for any observed event rate is no greater than 10 percent. In addition, the binomial probability of detecting three or more events is 88.2 percent when the true event rates are 5 percent or higher.
The study was designed jointly by the investigators and the program staff at the National Institute of Allergy and Infectious Diseases (NIAID) (the NIAID influenza team), and Dr. Treanor served as the principal investigator. The study was approved by the institutional review boards of the University of Rochester, University of Maryland, and the Los Angeles Biomedical Research Institute at Harbor–UCLA Medical Center. The vaccine product was manufactured by Sanofi Pasteur under contract to NIAID, but Sanofi had no role in the conduct of the study or the preparation of this report. The article was written jointly by the investigators. Dr. Treanor assumed responsibility for the writing and preparation of the manuscript and vouches for its accuracy and completeness.
Results
In the first stage of the study, 118 subjects received the first dose of vaccine or placebo, and 117 of these subjects received the second dose 28 days later. Enrollment in the first stage occurred in April 2005, with 28 subjects randomly assigned to receive 90 µg, 25 subjects assigned to 45 µg, 25 assigned to 15 µg, 28 assigned to 7.5 µg, and 12 assigned to placebo. One subject was unable to complete the study because of military service. After a review of the safety data by the independent data and safety monitoring board, the remaining 333 subjects were enrolled and received the first of the two doses. Enrollment in the second stage occurred in May 2005, and 75 subjects were randomly assigned to receive 90 µg, 73 were assigned to 45 µg, 76 were assigned to 15 µg, 73 were assigned to 7.5 µg, and 36 were assigned to placebo. Of these subjects, 13 did not receive the second dose, 11 because they were unable to make the follow-up visits, were unable to maintain compliance, or were ineligible and 2 because of adverse events. Two subjects were excluded from the immunogenicity analysis because they had participated in a previous study evaluating an H5 influenza vaccine. (A diagram of the disposition of the study subjects is available in the Supplementary Appendix.)
The median age of the enrolled subjects was 39 years (range, 18 to 64), and 54 percent of subjects were female; 79 percent of subjects were white, 11 percent were Asian, and 8 percent were black. Race was self-reported. Of the subjects, 42 percent had received conventional influenza vaccine in the previous year. The subjects' ages and demographic characteristics were similar in each of the study groups, but there were more women in the group assigned to 15 µg (as shown in Table 1 in the Supplementary Appendix).
Safety Analysis
The rates of symptoms reported during the first seven days after administration of each dose of vaccine are shown in Figure 1. Generally, the vaccine was well tolerated at all doses, and 84 percent of all reported symptoms were graded as mild by the subjects. There was no indication that the frequency or severity of either local or systemic symptoms was greater after the second dose than after the first dose, and there were no instances of anaphylaxis, hives, or other serious allergic reactions.
|
Systemic symptoms were relatively less common after either dose in all study groups and were not dependent on the dose; the frequencies of reports of feverishness, malaise, myalgia, headache, and nausea in all groups did not differ significantly from those in the placebo group (P>0.05). Eleven subjects reported fever (temperature, 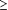 37.8°C; maximum, 38.2°C) after vaccination: 9 after the first dose (2 in the placebo group, 1 in the 7.5-µg group, 3 in the 15-µg group, and 3 in the 90-µg group) and 2 subjects after the second dose (1 in the placebo group and 1 in the 45-µg group). Clinical laboratory safety testing of subjects during the first stage did not reveal clinically significant abnormalities (described in the Supplementary Appendix).
37.8°C; maximum, 38.2°C) after vaccination: 9 after the first dose (2 in the placebo group, 1 in the 7.5-µg group, 3 in the 15-µg group, and 3 in the 90-µg group) and 2 subjects after the second dose (1 in the placebo group and 1 in the 45-µg group). Clinical laboratory safety testing of subjects during the first stage did not reveal clinically significant abnormalities (described in the Supplementary Appendix).
In one vaccine recipient, a rash developed after receipt of the first dose, and the subject did not receive the second dose. This subject noticed a nonpruritic, maculopapular rash over the abdomen and upper arms bilaterally on day 5 after the first dose of vaccine, without involvement of the face, hands or feet, or mucous membranes. The rash faded and resolved completely by day 42. Because the cause of the rash was unclear, the investigator elected not to administer the second dose of vaccine. This subject had no history of reaction to influenza vaccine, including rash.
There was one serious adverse event in the study, but it was judged by investigators to be unrelated to vaccination. A 52-year-old man in the second stage of the study died 24 days after receipt of the first dose of 45 µg of vaccine. He had a history of alcohol abuse that had not been revealed on enrollment, and the subject was noted to be consuming alcohol heavily. Autopsy revealed marked steatosis of the liver, and the death was determined by the medical examiner to be due to chronic alcoholism. (Additional information on this event is included in the Supplementary Appendix.)
Immunogenicity Analysis
The results of immunogenicity testing with the use of both hemagglutination inhibition and microneutralization are shown in Table 1 and Figure 2. As expected, in the majority of subjects, antibody against the A/Vietnam/2004 virus was not detected by either method before immunization, although 15 subjects (3 percent) had a positive hemagglutination-inhibition test, and 12 (3 percent) had a positive microneutralization test. The reasons for these positive results are unknown, because none of the subjects reported exposures that would be likely to result in H5 virus infection, and preliminary analysis has not suggested any relationship between H5 antibody and antibody against conventional human influenza viruses.
|
|
The prespecified primary immunogenicity end point chosen for this study was the development of a microneutralization titer of 1:40 or greater after two doses of vaccine. Figure 2 shows the distribution of antibody titers according to the hemagglutination-inhibition assay (Figure 2A) and microneutralization assay (Figure 2B) after vaccination in each group categorized according to dose. Only in the group receiving the 90-µg dose was the primary end point reached by more than 50 percent of the recipients. In this group, 54 percent of the subjects (95 percent confidence interval, 43 to 64 percent) had microneutralization titers of 1:40 or greater and 58 percent (95 percent confidence interval, 47 to 67 percent) had hemagglutination-inhibition titers of 1:40 or greater. The frequency of both these end points in the group receiving the 90-µg dose was significantly greater than in the other vaccine groups (P<0.001). Microneutralization titers of 1:20 or greater were seen in 70 percent of the group receiving the 90-µg dose and 57 percent of the group receiving the 45-µg dose, but in only 32 percent of the group receiving the 15-µg dose and 25 percent of the group receiving the 7.5-µg dose.
Because the highly pathogenic wild-type influenza A/Vietnam/1203/2004 virus can be manipulated only under strict conditions of biocontainment, the majority of serologic tests used the antigenically identical but apathogenic influenza rgA/Vietnam/1203/2004xA/PR/8/34 vaccine virus. The ability of serum samples from this study to neutralize the wild-type virus was confirmed in a subgroup of 63 samples obtained on day 56 from randomly selected specimens representing a spectrum of antibody titers, which were weighted toward higher responses to the apathogenic vaccine virus and assayed against the wild-type virus. The agreement in the antibody titers assayed against the two viruses was good, with a Spearman's correlation coefficient of 0.74 (P<0.001). Of the 53 samples tested in which the titers of antibody against the vaccine virus were greater than 1:40, 51 also had titers of antibody against the wild-type virus of more than 1:40.
Discussion
This study demonstrates that is it possible to generate immunity against H5 influenza with the use of a purified, subvirion vaccine administered in two relatively high doses. Our results are similar to those observed in a study conducted with the use of a purified, recombinant H5 hemagglutinin in humans,14 in which intramuscular administration of two doses of approximately 90 µg each of a baculovirus-expressed recombinant H5 hemagglutinin resulted in neutralizing antibody titers of 1:80 or greater in 56 percent of healthy adult recipients, whereas lower doses were considerably less immunogenic. Although that study used a different vaccine and slightly different assays to measure immune responses, both those results and ours show that high doses of relatively purified protein vaccines were required to induce immunity in most recipients.
The interpretation of the significance of these findings should be done in the context of our current understanding of immunity to H5 viruses. Our decision to use a neutralizing antibody titer of 1:40 as the primary immunogenicity end point was not based on observations of antibody-mediated protection in the field but, rather, on the development of criteria that appeared to distinguish between infected and uninfected persons during serologic surveys performed during the previous outbreak of H5 virus infection in 1997.15 It is possible that lower titers of neutralizing antibody could be associated with protection, as has been observed in studies of conventional human influenza viruses.16,17,18,19 The development of a sensitive hemagglutination-inhibition assay for H5 viruses12 should facilitate further studies, but the level of antibody associated with protection in this assay has yet to be determined.
On the basis of these preliminary data, a two-dose schedule of 90 µg of subvirion H5 vaccine would probably have an acceptable tolerability profile and could be effective in preventing H5 influenza in healthy adult recipients. Elderly persons, persons with impaired immunity, or children may have a different response, and trials of the vaccine in these populations are in progress. Production of the vaccine and this clinical trial are important steps toward control of a pandemic, and the current vaccine would probably be acceptable for licensure, if needed. However, the need for a vaccine with a total dose of 180 µg would pose a considerable barrier to rapid production of a supply that would be adequate to meet the world's requirements should a pandemic occur. Therefore, dose-sparing approaches should be pursued aggressively. These approaches could include the use of adjuvants such as aluminum20 or MF5921 and the use of intradermal administration of vaccine,22,23 both of which have been reported to be potentially dose sparing for influenza vaccines in small studies. In addition, the recent demonstration of a substantial increase in the immune response when a third dose of H5 vaccine was administered to subjects 16 months after a primary series24 suggests that another strategy for improving the immune response would be prepriming, perhaps by including an H5 component in the annual vaccine. Combinations of these approaches may be needed. Finally, live attenuated vaccines are being developed. As the results of studies to evaluate each of these options become available, our results may be useful for comparison.
Supported by contracts with the National Institute of Allergy and Infectious Diseases (N01 AI 25460 to Dr. Treanor, AI 25461 to Dr. Campbell, AI 25463 to Dr. Zangwill, and AI 30068 to Dr. Rowe) and grants from the federal General Clinical Research Center (M01 RR00044 to Dr. Treanor, M01 RR165001 to Dr. Campbell, and M01-RR00425 to Dr. Zangwill). None of the authors report having received financial support from Sanofi Pasteur. Dr. Treanor reports having received or applying for grant support from Merck, Protein Sciences, VaxGen, AlphaVax, ID Biomedical, EpiVax, and PowderMed. No other potential conflict of interest relevant to this article was reported.
We are indebted to the many subinvestigators, study nurses, and technicians who contributed to this study, including: C. Mhorag Hay, Diane O'Brien, and Carrie Nolan (University of Rochester); Wilbur Chen and Mary Lou Mullen (University of Maryland Center for Vaccine Development); Midi Mikasa, Pat Chatfield, Sacred Cartwright, Susan Partridge, Elizabeth Moore, Swei-ju Chang, Rita Manai, and Merlyn Dubria (Los Angeles Biomedical Research Institute); Michael McDowell, Lisa Slappey, Franziska Rosser, Diana Noah, Lucile White, and Mindy Sosa (Southern Research Institute); Fred Batzold (Clinical Trials Management, Division of Microbiology and Infectious Diseases, National Institutes of Health); John Coleman, Erich Hoffman, and Richard Webby (St. Jude Children's Research Hospital); Richard Hjorth, Desirée Taylor, and Charles Whittaker (Sanofi Pasteur); Heather Hill, Jill Kissel, Dewei She, Bernadette Jolles, and Ken Wilkins (EMMES); and Jean Hu-Primmer, Emily Kough, Linda Lambert, Pamela McInnes, Katherine Muth, and Shy Shorer (National Institute of Allergy and Infectious Diseases Influenza Team).
Source Information
From the Department of Medicine, University of Rochester, Rochester, N.Y.(J.J.T.); the Center for Vaccine Development, University of Maryland School of Medicine, Baltimore (J.D.C.); the Los Angeles Biomedical Research Institute and UCLA Center for Vaccine Research, Harbor–UCLA Medical Center, Los Angeles (K.M.Z.); Southern Research Institute, Birmingham, Ala. (T.R.); and EMMES, Rockville, Md. (M.W.).
Address reprint requests to Dr. Treanor at the Department of Medicine, University of Rochester Medical Center, 601 Elmwood Ave., Rm. 3-6309, Rochester, NY 14642, or at john_treanor{at}urmc.rochester.edu.
References
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
Related Letters:
An Inactivated Subvirion Influenza A (H5N1) Vaccine
Johansson B. E., Brett I. C., Focosi D., Poland G. A.
Extract |
Full Text |
PDF
N Engl J Med 2006;
354:2724-2725, Jun 22, 2006.
Correspondence
This article has been cited by other articles:
HOME | SUBSCRIBE | SEARCH | CURRENT ISSUE | PAST ISSUES | COLLECTIONS | PRIVACY | HELP | beta.nejm.org Comments and questions? Please contact us. The New England Journal of Medicine is owned, published, and copyrighted © 2009 Massachusetts Medical Society. All rights reserved. |

