


Center for Biologics Evaluation and Research
FY 2006 Annual Report
Innovative Technology Advancing Public Health
Note: Documents in PDF format require the Adobe Acrobat Reader®. If you experience problems with PDF documents, please download the latest version of the Reader®.
Table of Contents
- Blood and Blood Products
- Vaccines and Vaccine Safety
- Cellular and Gene Therapies
- Tissues
- Xenotransplantation
- Devices
- Allergenics
National Preparedness And Security
- Pandemic Influenza Preparedness
- Vaccine Production and Supply
- Ensuring a Safe and Available Blood Supply
- Protecting America from Terrorism
- Anthrax
- Smallpox
- Blood Supply
- Other Counterterrorism Activities
- Improving the Safety of the Blood Supply with Novel Donor Screening Tests
- Hepatitis B
- West Nile Virus
- Monitoring the Availability of Blood and Blood Components
- Enhancing the Safety of Blood and Development of the CBER Blood Safety Team
- Biological Safety Activities
- CBER Outreach Update
Increase Access To Innovative Products And Technologies To Improve Health
- Initiatives to Strengthen, Diversify, and Increase Capacity for Influenza Vaccines
- The 2005-2006 Influenza Season
- FDA Issues Advice to Facilitate Influenza Vaccine Development
- Technology for New-Generation Vaccines for Viral Infectious Diseases
- Critical Path Initiative: Personalized Medicine
- Research Management Advances
- Scientific Expertise Teams
- Scientific Training Initiatives
- PDUFA
- MDUFMA
- Cellular and Gene Therapies: Facilitating Availability and Development of Safe and Effective Products through New Technologies
- Genomics and Proteomics
- Tissue Engineering
- Cellular and Gene Therapies: Outreach and Partnerships
- FDA Approves Gardasil
- FDA Approves Zostavax
- FDA Approves RotaTeq
- FDA Approves Vivaglobin
- FDA Approves New Manufacturer for Cytogam
- FDA Approves ADVIA Centaur HIV 1/O/2 Assay A
Improve Product Quality, Safety, And Availability Through Better Manufacturing And Product Oversight
- Ensuring Safety of Recipients of Donor Tissue
- Donor Referral Services
- Immune Globulin Availability and Demand
- TSE Safety for Biological Products
- Blood Donor Deferral for Transfusion in France
- TSE Research and Risk Estimation
- Workshop on Testing for Malarial Infections in Blood Donors
Transform Administrative Systems And Infrastructure To Support FDA Operations
- Globalization of Public Health and Product Development: International Activities
- Highlights
- WHO and PAHO Activities
- Global Collaboration on Blood Safety
- International Conference on Harmonisation
- International Partnering
- Pharmaceutical Inspection Cooperation Scheme (PIC/S)
- Global Harmonisation Task Force
- International Outreach
- Review Management Initiatives
- Agency Harmonisation Initiatives
- Quality Systems Initiatives
- Electronic Standards Initiatives
- Outreach Initiatives
- CBER Review Business Process Efforts
- Quality Assurance
- Management Systems Initiatives
- IT and System Enhancements
- eSystems
- EDR and FDA Gateway
- Other System Enhancements
- Security
Appendix A (CBER Publications)
Appendix B (CBER Exhibit Program - FY 2006)
Appendix C (CBER Major Approvals - FY 2006)
Appendix D (Rulemaking and Guidance Documents - FY 2006)
Appendix E (Advisory Committee Meetings - FY 2006)
Appendix F (Organizational Chart)
Vision and Mission
Vision
The Center for Biologics Evaluation and Research (CBER) uses sound science and regulatory expertise to:
- protect and improve public and individual health in the United States (U.S.) and globally
- facilitate the development of, approval of, and access to safe and effective products and promising new technologies
- strengthen CBER as a pre-eminent regulatory organization for biologics.
Mission
To ensure the safety, purity, potency, and effectiveness of biological products, including vaccines; blood and blood products; and cells, tissues, and gene therapies for the prevention, diagnosis, and treatment of human diseases, conditions, or injuries. Through our mission, we also help to defend the public against the threats of emerging infectious diseases and bioterrorism.
In fulfilling our mission as a center in the U.S. Food and Drug Administration (FDA), we apply the following principles with the highest ethical standards and integrity:
- develop, maintain, and support a high-quality and diverse workforce
- ensure compliance with laws and regulations through review, education, surveillance, and enforcement
- conduct research as an essential element of science-based decision-making.
A Message from the Director
July 2007
Dear Colleagues in the Biologics Community:
It is my pleasure to share the tenth annual report from the Food and Drug Administration's (FDA's) Center for Biologics Evaluation and Research (CBER). This report focuses on accomplishments and on-going initiatives during Fiscal Year 2006 (FY 2006). CBER's products touch people's lives on a daily basis and they play a critical role in the nation's public health preparedness, where they are critical in addressing threats from emerging infectious diseases, like pandemic influenza, and terrorism. They include a wide range of both essential and highly innovative products, including vaccines and allergenic products, blood and blood products, tests used in assuring the safety of transfusion, and human tissues, cell and gene therapies. The goals of the Center align with those established by the FDA to accomplish our mission and protect public health. CBER's FY 2006 annual report is organized within the FDA's four goal areas and highlights accomplishments in the area of Public Health Preparedness and Security. CBER works closely with multiple partners to achieve the following goals:
- Enhance Patient and Consumer Protection and Empower Them with Better Information about Regulated Products
- Increase Access to Innovative Products and Technologies to Improve Health
- Improve Product Quality, Safety, and Availability through Better Manufacturing and Product Oversight
- Transform Administrative Systems and Infrastructure to Support FDA Operations.
In each of these areas, as well as in the area of Public Health Preparedness and Security, CBER has made substantial accomplishments and exceeded goals. We continue to meet or exceed the user fee performance goals in the Prescription Drug User Fee Act (PDUFA III), and the Medical Device User Fee and Modernization Act (MDUFMA). Our success in accomplishing thorough but timely scientific review of biological products and related devices has resulted in safe and effective products reaching those in need more efficiently and rapidly. CBER's interactions with applicants, our application review efforts, and proactive monitoring and transparency regarding product quality and safety issues, have helped to keep needed vaccines, tissues and blood products both safe and available, thus maintaining public confidence and trust.
Safety expectations for blood, tissues and vaccines are particularly high; and product availability, critical for our nation's health and medical infrastructure, is also frequently a challenge. Our product safety activities emphasize effective, interdisciplinary, proactive approaches using modern methods to prevent, detect, and promptly investigate potential problems, and assure needed supplies, while providing effective and transparent communication to consumers and health professionals. Among our recent safety innovations and accomplishments have been the implementation of a new regulatory framework to provide enhanced oversight of tissue safety and the creation of formal interdisciplinary product safety teams (e.g. the Tissue Safety Team and the Blood Safety Team). These teams meet regularly and in emergencies to bring together diverse disciplines (e.g. product scientists, clinicians, epidemiologists, manufacturing experts, and communications experts) to share, analyze and act on safety information and develop needed strategic and scientific approaches to enhance safety. The safety teams collaborate with other FDA components and colleagues at other agencies, particularly the Centers for Disease Control and Prevention (CDC). In addition, we have pioneered the use of large medical databases to detect and investigate product safety issues. We have also pioneered the use of risk assessment computer modeling to evaluate risk and help inform patients and health care providers of effective approaches to help reduce risk (e.g., an assessment of the potential risk from the agent of variant Creutzfeld-Jacob disease, the cause of "Mad Cow" disease, to blood plasma product safety).
To address the global threat of pandemic influenza, and position us to better respond to other future emerging threats, CBER has taken a proactive leadership role and worked with the World Health Organization (WHO), the Department of Health and Human Services (DHHS), other Federal agencies, particularly CDC and the National Institutes of Health (NIH), industry, and sister regulatory authorities from other nations. We have acted with these partners to foster information sharing, rapid development, evaluation and U.S. and global availability of pandemic influenza vaccines that will be effective, safe and high quality. We conducted outreach to major manufacturers throughout the world to stimulate interest in producing vaccines for the U.S. market for both annual and pandemic influenza. We have taken a number of steps aimed at enhancing manufacturers' capacity to produce more doses every year of annual vaccine. As a result, the United States has advanced from a vaccine supply of 3 manufacturers and approximately 63 million doses in the 2004-2005 season to 5 manufacturers and 120 million doses in the 2006-2007 season. We also co-sponsored a series of meetings with vaccine regulators from around the world to establish agreed upon standards for human avian influenza vaccines and have published guidances, including provisions for accelerated approval based on immunogenicity, to speed and assist manufacturers' development of vaccines for both seasonal and pandemic influenza. We are also strengthening our emergency response infrastructure and processes (e.g. IT, communications) and our laboratory testing, standards and methods activities, including use of quality systems approaches. This strengthening will allow us to be better prepared with the tools and capacity we, and industry, will need to help assure vaccine and other critical product availability and quality should a pandemic occur.
Our international activities have become increasingly critical and have had growing impacts as emerging health threats, product manufacturing and use, and innovation and advances have all become more prevalent. CBER is highly engaged in global information sharing, capacity building and harmonisation to benefit global health. We actively participate in the Regulatory Preparedness Workshops on Human Vaccines for Pandemic Influenza. These workshops aid in the development of guidelines and standards for global regulatory information sharing, coordination and pandemic vaccine evaluation and quality and have provided input to the WHO for the development of, "WHO Guidelines on Regulatory Preparedness for Human Pandemic Influenza Vaccines." Additionally, we have engaged in regulatory outreach, with respect to avian influenza preparedness, to our counterparts in Asia. This is just a snapshot of our commitment to protecting and promoting global health and addressing major neglected diseases such as tuberculosis, HIV, and malaria, which is essential to improve lives throughout the world and protect our nation and its people. We do this through a large inventory of activities that range from helping to build regulatory capacity in developing countries, collaborating with WHO and other partners on sound global public health policy and working with industry, NIH, nongovernmental organizations and others to promote the development of safe and effective products to address unmet global needs.
Through the advancement of new technologies, we have helped make available a number of products that are both the first of their kind and represent significant advances in public health. We approved a human papillomavirus vaccine to prevent cervical cancer and precancerous lesions caused by certain human papillomaviruses. We approved several new blood tests that help make the blood supply even safer, such as a test to detect West Nile virus infection, a fully automated diagnostic test for antibodies to HIV-1, HIV-2, and HIV-I Group O in human serum and plasma, and a fully automated product combining screening and confirmatory testing for hepatitis B (HBsAg). CBER co-developed the Blood Availability and Safety Inventory System (BASIS) used by DHHS to monitor the availability of blood components at major hospital centers and blood establishments throughout the United States. We published a new guidance on umbilical cord blood, increasingly used in treatment of diseases such as cancer and leukemia, which is designed to foster the availability of these stem cells, while assuring their effectiveness and safety.
With dedication, hard work, and team effort from our remarkably diverse and knowledgeable staff, CBER continues to achieve outstanding accomplishments in promoting innovation while ensuring the quality, safety and availability of the products we regulate. We strive for continuous quality improvement, including enhanced efficiency. Each of us at CBER is committed to our mission and we all appreciate your essential input, support and participation in achieving our goals. I welcome your feedback and ideas to strengthen the mission of the Center, and our common public health goals, and look forward to working with you in the coming year.
Jesse L. Goodman, M.D., M.P.H.
Director
Center for Biologics Evaluation and Research
Our Products
Blood and Blood Products*
FDA is responsible for ensuring the safety of the nation's blood supply by minimizing the risk of infectious disease transmission and other hazards, while facilitating the maintenance of an adequate supply of blood and blood products. CBER regulates blood and blood components that are used for transfusion and for manufacture into products such as plasma derivatives. CBER also regulates the resulting blood products, such as clotting factors, concentrates, immune globulins, albumin and protease inhibitors. CBER also establishes product standards and performs lot release testing. CBER regulates devices used to prepare blood products, including blood establishment computer software (BECS), cell separators, and blood collection containers, as well as tests to screen blood donors for human immunodeficiency virus-type 1 (HIV1) and other viruses, such as hepatitis B virus (HBV) and hepatitis C virus (HCV), West Nile virus (WNV), human T-lymphotropic virus types I and II (HTLVI/II), and for syphilis. CBER develops and enforces quality standards, monitors, analyzes, and acts on reports of biological product deviations and adverse clinical events. Biological product deviations are the result of unexpected or unforeseeable events in the manufacturing process.
For example, FDA encouraged development of highly sensitive nucleic acid tests (NATs) for the detection of HIV-1 and HCV. These tests are now FDA-approved and recommended for use in screening blood donors to reduce the risk of transmission of these infectious agents. To standardize the performance of these tests, CBER developed essential lot release reagents. FDA also encouraged the development of NATs for WNV in response to the epidemic in the U.S. Close coordination with other U.S. Public Health Service (PHS) agencies, device manufacturers and blood establishments resulted in the development and use of investigational WNV tests in blood establishments within 8 months of recognition of this new transfusion transmission risk. As a result of these efforts, FDA approved a WNV NAT for use in blood screening within the goal timeframe established for licensure of the test.
Over a period of years, FDA has progressively strengthened the overlapping safeguards that protect patients from unsuitable blood and blood products. Blood donors are asked specific questions and review educational materials about risk factors that could indicate possible infection with a transmissible disease. This upfront donor screening helps to reduce risk from infectious agents by identifying and deferring potentially high-risk donors prior to testing and is especially important in the absence of donor screening tests for emerging infectious diseases. CBER has worked actively with the blood community over the past decade to redesign, streamline and cognitively assess a standardized donor-screening questionnaire. This questionnaire now is in broad use throughout the U.S. for the screening of blood donors.
FDA facilitates the development and implementation of sensitive tests to detect infectious agents in blood. Testing of donors for infectious agents is a critical safeguard for blood safety. To further enhance blood safety, FDA requires blood centers to maintain lists of donors considered unsuitable to donate blood because of test results or risk factors for infectious diseases that can be transmitted by blood. Blood establishments also quarantine units of blood until they determine the units are suitable for release. In addition, FDA has significantly increased its oversight of inspections of blood collection and manufacturing facilities.
*See Appendix A References 1-6
Vaccines and Vaccine Safety*
CBER regulates vaccine products. Many of these products are pediatric vaccines that have contributed to the dramatic reduction or elimination of life-threatening childhood diseases in the U.S. (e.g., diphtheria, measles, and polio). Newer vaccines are playing an increasing role in protecting and improving the lives of adolescents and adults and include vaccines to prevent meningococcal disease, shingles, and cervical cancer. In addition, there are vaccines under development that offer the promise of preventing serious infectious diseases, such as pandemic influenza viruses and severe acute respiratory syndrome (SARS), HIV-1, and malaria. As with all medical products, highly-trained scientists and clinicians rigorously review laboratory and clinical data in assessing the safety, effectiveness, and quality of vaccines. FDA also reviews additional studies after some vaccines are approved to further evaluate their safety and effectiveness (e.g., in broader population groups). Both before and after a vaccine is licensed, FDA inspects vaccine manufacturing facilities to help ensure continued high quality and safe production.
The U.S. Centers for Disease Control and Prevention (CDC) and CBER jointly manage the Vaccine Adverse Event Reporting System (VAERS), a cooperative program for vaccine safety. VAERS is a postmarketing safety surveillance program that collects information about adverse events (side effects) potentially related to vaccination and reported after the administration of U.S. licensed vaccines. In collaboration with CDC, state health departments, and other partners, CBER uses VAERS to monitor vaccine adverse event reports for possible indicators of vaccine safety concerns.
*See Appendix A References 7-15
Cellular and Gene Therapies*
CBER regulates cellular and gene therapy products including therapeutic cancer vaccines. Somatic cells, vectors expressing certain gene products, and genetically manipulated cells offer the promise of harnessing the power of different cell types to fight disease, restore normal function, repair injuries, replace lost cells, or regenerate failing organs. CBER is aware of both the promise of gene therapy and its potential to cause serious adverse events and works closely with the National Institutes of Health (NIH), academia, and industry on these products. For example, FDA and NIH have collaboratively developed a Web-accessible database on human gene transfer—the Genetic Modification Clinical Research Information System (GeMCRIS). GeMCRIS enables faster reporting of adverse events in human gene transfer trials and is a unique public information resource. The system provides information to the public directly via the Internet at www.gemcris.od.nih.gov and improves the government's ability to monitor adverse events in gene therapy. Manufacturers of gene and cellular therapy products must study their products in the laboratory for safety before beginning studies in humans under an Investigational New Drug Application(IND) or an Investigational Device Exemption (IDE). Like all biological products, gene and cellular therapies need to meet statutory and regulatory requirements for safety, purity, and potency before the products can be licensed for commercial distribution in the U.S. Through FY 2006, CBER has received 517 gene therapy INDs and is currently overseeing approximately 270 active INDs. CBER has also received 1,171 INDs and IDEs for somatic cellular therapies through FY 2006, and approximately 437 are still active. There have been a total of 40 INDs and IDEs for xenotransplantation products, and approximately 14 of them were active at the end of FY 2006.
CBER has provided proactive scientific and regulatory advice to manufacturers in areas of novel product development. Focusing on how best to evaluate essential issues of safety and efficacy, while facilitating product development, we are also committed to protecting human study subjects. Our involvement in broad public interactions helps CBER and product developers address important issues involving the development of novel gene and cellular therapy products.
Tissues* Tissue transplantation is a rapidly growing industry. The number of musculoskeletal tissue transplants increased from approximately 350,000 in 1990 to more than 1.6 million in 2006. CBER is responsible for regulating many different types of human tissue and cells that are transplanted during various types of medical procedures (e.g., skin replacement following severe burns, tendons and ligaments used to repair injuries, bone replacement, and corneas used to restore eyesight). Transplantation of human tissues presents unique safety challenges, in particular the risks of transmitting infectious diseases from donor to recipient and the contamination of tissues during processing. These risks can be reduced significantly, but not completely eliminated. Since 1993, CBER has also required tissue establishments to screen and test donors. Since 1997, CBER has required tissue establishments to prepare, validate, and follow written procedures to prevent contamination and cross-contamination during processing. In response to the increased use, role, and complexity of tissue transplants, and the recognition of threats to tissue safety, FDA developed and has implemented a comprehensive new framework, which went into effect in May 2005, for the regulation of human cells, tissues, and cellular- and tissue-based products. The new framework promotes the use of the most up-to-date tools and methods to reduce risks of infectious disease transmission and contamination. FDA's regulatory framework includes a broad range of tissues (e.g., hematopoietic stem cells and reproductive tissues, the latter covered primarily for donor eligibility and testing issues). The new regulations encourage a comprehensive, yet flexible, approach to quality throughout the entire manufacturing process, from donor assessment to the final product, including adverse event reporting. CBER conducted extensive outreach and sought stakeholder input as we developed the regulatory approach, and we continue to seek input in this important area. |
 |
On August 30, 2006, the FDA established a multidisciplinary task force on human cell and tissue safety. The FDA Human Tissue Task Force (HTTF) was established as part of the Agency's efforts to strengthen its comprehensive, risk-based system for regulating human cells and tissue. The main priority of HTTF is to assess the effectiveness of new tissue regulations, and conduct a review of recent reported findings that some tissue recovery establishments are not following federal requirements for tissue recovery. The HTTF is led by senior FDA officials from CBER and the Office of Regulatory Affairs.
Xenotransplantation*
CBER regulates products used in xenotransplantation, which is any procedure that involves the transplantation, implantation, or infusion into a human recipient of either: (1) live cells, tissues, or organs from a nonhuman animal source; or (2) human body fluids, cells, tissues, or organs that have had contact with live, nonhuman animal cells, tissues, or organs. Xenotransplantation offers the promise of providing needed organs and tissues to thousands of individuals who await transplants of scarce human organs. It holds the potential for the treatment of a wide range of conditions and disorders, including diabetes, degenerative neurological disorders, and other diseases involving tissue destruction and organ failure. Currently, the demand for human organs for clinical transplantation far exceeds the supply. Although xenotransplantation's potential benefits are considerable, it raises a number of complex scientific and public health challenges, most notably the risk of transmission of infectious diseases from animals to humans and the failure of such transplants due to rejection. CBER's continued careful oversight is critical to protecting public health while exploring the potential of these investigational therapies. CBER continues to be a leader in international activities aimed at the safety and regulation of xenotransplantation products.
Devices
CBER regulates many medical devices used in the collection, processing, testing, manufacturing, and administration of blood, blood components, human cells, tissues, and cellular- and tissue-based products. The Center's activities include regulation of HIV-1 and other infectious disease test kits used to screen donor blood, blood components, and cellular- and tissue-based products. CBER also regulates HIV-1 tests used to diagnose, treat, and monitor therapy in persons infected with HIV. CBER collaborates closely with FDA's Center for Devices and Radiological Health (CDRH) on cross cutting issues. We also work with CDRH and the Office of Combination Products in the regulation of combination products, such as tissue-engineering products. The Center has also leveraged its resources by working with the National Toxicology Program (NTP), a joint FDA-NIH venture, to evaluate safety issues associated with materials used in blood collection and transfusion devices.
*See Appendix A References 18-20
Allergenics*
Licensed allergenic products include: (1) allergen patch tests, and (2) allergenic extracts. Allergen patch tests are diagnostic tests applied to the surface of the skin. Patch tests are used by physicians to determine the specific causes of contact dermatitis, and are manufactured from natural substances or chemicals (nickel, rubber, and fragrance mixes) that are known to cause contact dermatitis. Allergenic extracts are used for the diagnosis and treatment of allergic diseases, such as allergic rhinitis (or hay fever), allergic sinusitis, allergic conjunctivitis, bee venom allergy, and food allergy. CBER has been proactive in evaluating novel technological approaches for improving allergenic product development and standardization, as well as characterizing these complex biological products. Some allergenic extracts are currently standardized, whereas others are nonstandardized. Standardized allergenic extracts are compared to U.S. reference standards for potency before release. CBER maintains these reference standards and distributes them to manufacturers. There are currently 19 standardized allergenic extracts.
*See Appendix A References 21-24
National Preparedness And Security
CBER plays an important role in protecting America from terrorism. A major focus for the Center is the expeditious development and licensing of products to diagnose, treat, or prevent disease from exposure to the pathogens that have been identified as bioterror threats. CBER plays a crucial role in protecting public health by ensuring the availability of safe and effective medical countermeasures for mitigating the consequences of a pandemic, natural, or deliberate disaster. The Center also works to ensure the continuity of mission critical operations in the face of disasters.
Pandemic Influenza Preparedness*
Vaccine Production and Supply
Pandemic influenza is a significant public health threat to our nation and the world. The supplemental funding that CBER received in 2006 allowed us to significantly increase the resources that we direct to the review and development of guidance for influenza vaccines, and the research that supports these activities. Around the globe, different nations are developing plans to prepare for, and respond to, the next influenza pandemic. Although no influenza pandemic has occurred in the U.S. in decades, scientists are concerned that the highly pathogenic avian influenza virus (H5N1), which is currently circulating in wild and domestic birds in Asia, Europe, the Middle East, and Africa, and has caused deaths in humans, may mutate into a form capable of efficient and sustained human-to-human transmission, thereby resulting in a global outbreak (or pandemic). Preparedness planning is imperative to lessen the impact of such a pandemic.
CBER is engaged in a leadership role to prepare for and respond to the risks of a pandemic influenza outbreak. To safeguard Americans from the danger of pandemic influenza, CBER is working with industry, with other agencies in the Department of Health and Human Services (DHHS), and global partners to facilitate the development and availability of pandemic influenza vaccines in the shortest time possible to protect the largest number of people using a vaccine that is safe, effective, and easy to deliver.
As a result of our intense efforts to help build manufacturing capacity for pandemic and seasonal influenza vaccines, CBER has made many strides in aiding vaccine manufacturers. In March 2006, the Agency published two draft guidances outlining specific approaches that influenza vaccine developers can follow to show the safety and effectiveness of new seasonal and pandemic influenza vaccines in support of licensure. These guidances also provide flexible regulatory pathways for getting influenza vaccines on the market, including an accelerated approval process that can substantially reduce the development time for a new vaccine.
Currently, influenza vaccines licensed in the U.S. are produced using fertile hens' eggs as part of a complex and exacting process. These guidances also apply to vaccines made with new manufacturing methods, such as cell culture-based and recombinant technologies, and adjuvants (substances that may enhance the body's immune response to a vaccine, making the vaccine more effective). In September 2006, CBER issued draft guidance on the production of viral vaccines in cell cultures, which is directly applicable to, and should help facilitate, the development of cell-culture based pandemic and seasonal influenza virus vaccines.
Responding to the threat of a pandemic strain of influenza will require an expedited evaluation and licensure of new vaccines to ensure adequate supplies of influenza vaccine during the pandemic period. CBER's scientific research program contributes to pandemic influenza preparedness by facilitating the availability of influenza vaccine. CBER is involved in the following activities:
- preparing strain-specific antisera that are used to determine the potency of vaccines against a pandemic strain
- examining the genetic makeup of potential high-growth viruses to develop reference vaccine viruses that grow optimally in either eggs or cell cultures
- exploring what is needed to prepare libraries of potential pandemic influenza viral strains
that could be readily available to begin manufacturing vaccines.
CBER also collaborates to develop safety tests that help evaluate whether cell cultures that have the potential for tumorigenicity can be used to manufacture vaccines. Activities also included in the development of assays to detect infectious agents that could potentially contaminate cell cultures. In addition, the Division of Product Quality, created in the Office of Vaccines Research and Review, has the responsibility for facilitating the lot release testing for influenza vaccines. By doing this work now, we hope to have the necessary tools in hand to respond to a pandemic before it occurs.
Seasonal influenza vaccine manufacturers will likely produce pandemic influenza vaccines, therefore increases in manufacturing capacity and improvements to existing processes could enhance the ability to produce pandemic influenza vaccines. CBER uses knowledge gained from past experience with counterterrorism products and with the 2004-2005 influenza vaccine shortage to facilitate the regulatory review of new Biologic License Applications (BLA) and supplements of existing applications for influenza vaccines. In response to the 2004 influenza vaccine shortfall, FDA contacted major manufacturers of influenza vaccine throughout the world to stimulate interest in producing vaccine for the U.S. market, including the provision of an accelerated approval pathway. As a result, licensed influenza vaccines have risen from three to five within a two-year period. Having additional manufacturers enhances the capacity to produce more doses of seasonal influenza vaccine every year, provides better protection against future interruptions in supply, and enhances the manufacturing capacity to produce pandemic influenza vaccines.
CBER is working with multiple partners, including manufacturers, to transform the influenza marketplace, reinvigorate influenza vaccine infrastructure through promising new technologies, secure additional licensed vaccines and medicines, and prepare stronger response plans and capacity. With the goal of increasing access to safe and effective pandemic vaccines worldwide, CBER has been working with the World Health Organization (WHO) and foreign regulatory counterparts to develop international convergence with regard to the scientific and regulatory standards for safety, effectiveness, and manufacturing quality of pandemic influenza vaccines. Together with the WHO and Health Canada, CBER co-sponsored two workshops, one in Canada in March 2006, and the second in the U.S. in June 2006, where regulatory authorities from developed and developing nations provided input that formed the basis for WHO's draft guidelines for pandemic influenza. This draft, together with comments from manufacturers, will be further discussed at a workshop in Geneva in 2007.
Ensuring a Safe and Available Blood Supply
At CBER, preparedness for pandemic influenza also includes ensuring a safe and available blood supply. We are carefully developing our own internal Continuity of Operations Plan for pandemic influenza, as well as coordinating with other federal agencies and national organizations. Key partners in this effort include the DHHS, Office of the Secretary and Public Health Science (OSPHS), CDC, and the American Association of Blood Banks (AABB). These collaborative activities are a critical and continuous part of CBER's pandemic influenza preparedness. To ensure timely communication and information sharing, CBER staff led the DHHS/PHS Pandemic Influenza Working Group that shares information and develops pandemic influenza preparations related to the blood supply. In addition, CBER leads a monthly PHS blood meeting that includes pandemic planning as a standard topic. The Center participates actively as a liaison with the AABB Disaster Task Force and the AABB Pandemic Influenza Task Force, both of which are proactively planning responses to the threat of pandemic influenza.
Center staff are considering potential regulatory approaches to the prompt evaluation and approval of new therapeutics (e.g., hyperimmune globulins), and new in vitro diagnostics (e.g., an H5N1 blood donor screening assay) that might be submitted to FDA. Within FDA, CBER strives to coordinate a safe, effective, and balanced plan in the event of a public health crisis of pandemic influenza. CBER investigates the feasibility of using newer technologies to detect contamination of the blood supply with bioterror/biowarfare infectious agents.
*See Appendix A References 25-29
Protecting America from Terrorism*
CBER is responsible for helping to ensure safe and effective biological products are available for treating and preventing illness due to terrorist agents. These products include vaccines, blood and blood derivatives, gene therapies, and cells and tissues for transplantation.
These biological products are carefully reviewed, and risk-to-benefit issues are carefully considered throughout their development, manufacturing, and clinical testing. CBER staff guide the products through the regulatory process, including manufacturing, preclinical testing, clinical trials, and the approval and licensing processes. Experts from diverse areas help expedite the development, evaluation, and approval processes. Often, time is of the essence, and the scientific and product issues are extremely challenging. Early involvement by scientific, statistical/ epidemiological, and clinical review staff is crucial to the success of the expedited development and review processes.
As part of national policy, a high priority is placed on category A agents—a CDC designation given to the greatest public health threats. Category A agents include organisms that cause anthrax, plague, smallpox, tularemia, and viral hemorrhagic fevers, as well as botulinum toxin. Emergency response proficiency is also being addressed through reassessing and strengthening capabilities and the development of continuity of operations plans. In addition, CBER has been proactive in identifying gaps that exist and in medical countermeasures against biological agents.
Anthrax
Anthrax is an infectious disease caused by the spore-forming bacterium, Bacillus anthracis. There are three forms of anthrax infection: (1) cutaneous; (2) gastrointestinal; and (3) inhalational, the latter of which is associated with the highest death rates. Currently, there is one anthrax vaccine—BioThrax, manufactured by BioPort Corporation, that is licensed in the U.S. This vaccine is indicated for pre-exposure prophylaxis against Bacillus anthracis (the causative agent of anthrax) in individuals between 18 and 65 years of age who are at risk of exposure. CBER has approved supplements to the BLA for BioThrax to increase the manufacturing capacity and extend the dating period of the product to 36 months.
FDA also issued a Proposed Rule and Order regarding the safety and efficacy of certain bacterial vaccines and toxoids, including the licensed anthrax vaccine that was published December 29, 2004. After reviewing the comments submitted to the docket, CBER, together with other FDA components, issued a Final Rule and Order regarding the safety and efficacy of certain licensed biological products. A Final Order was issued on December 15, 2005 regarding the authorization of emergency use of anthrax vaccine.
CBER is part of an interagency working group—with NIH, CDC, the Department of Defense (DoD), and DHHS—focused on encouraging the development of new, recombinant anthrax vaccines intended to prevent anthrax both before and after exposure. Such vaccines, being developed under INDs, present many challenges, especially in terms of developing reproducible animal models for demonstrating efficacy. The genetic makeup of anthrax is being studied to help improve vaccines and treatments.
DHHS awarded the first contract under Project Bioshield for a recombinant protective antigen (rPA) anthrax vaccine. CBER has devoted extensive resources to assist and guide manufacturers through the regulatory process in the development of their rPA anthrax vaccines and has provided extensive technical input to the Office of Public Health Emergency Medical Countermeasures (OPHEMC) in the Office of the Assistant Secretary for Public Health and Emergency Preparedness (OASPHEP), DHHS. New immune-based therapies for treating anthrax are also under development. For example, anthrax immune globulin is under evaluation as a potential treatment of anthrax disease. CBER has been working with the Center for Drug Evaluation and Research (CDER) to develop a draft guidance that addresses the development and licensure of immune-based therapies to treat anthrax disease. CBER has also provided technical input to DHHS the Strategic National Stockpile.
Smallpox
Smallpox, caused by the variola virus, is highly contagious and can be spread by close contact with an individual who has smallpox symptoms: high fever, fatigue, headaches, backaches, vomiting, rash, and pus-filled blisters. There is no proven treatment. The last confirmed case of smallpox in the U.S. was in 1949, and the last naturally occurring case in the world was recorded in Somalia in 1977. The death rate in the past was about 30% for the general population, and death rates can be higher for infants and young children.
Smallpox can be prevented through vaccination. Dryvax (dried, calf lymph type smallpox vaccine), made by Wyeth Laboratories, is the only smallpox vaccine currently licensed and is no longer being manufactured. Smallpox vaccines that are related to the same vaccine strain used in Dryvax, but grown in cell culture, are being developed. CBER is studying the safety, efficacy and manufacture of smallpox vaccines to facilitate regulation of these important products.
Blood Supply
Any time there are large emergencies or outbreaks of diseases, the blood supply is threatened. In the case of mass vaccinations, those who receive some vaccinations containing live viruses cannot be blood donors for a period of time because of the potential to transmit the vaccine virus. The Center has issued guidances on reducing the risk of transmitting diseases through blood donated by infected individuals, either by vaccination (anthrax and smallpox) or by exposure to a bioterrorist agent (bacilius anthracis). Recommendations have also been made for national emergency planning to ensure that vaccination campaigns consider impacts on the blood supply. Efforts are also underway to produce diagnostic assays to detect bioterrorist agents in blood donations.
CBER works in close liaison with the AABB-sponsored Interorganizational Task Force on Domestic Disasters and Acts of Terrorism. This task force defines a disaster or act of terrorism as an event that requires a much larger amount of blood than usual; one that temporarily restricts blood collection, testing, and distribution; or one that creates a sudden influx of donors requiring accelerated drawing of blood. CBER also works with multiple other partners to help ensure blood donations remain safe and plentiful in times of disaster.
Other Counterterrorism Activities
CBER continues to interact intensively with DHHS, the Department of Homeland Security (DHS), DoD, and industry on a broad array of projects to help make our nation better prepared for threats of biological, chemical, and radiological/nuclear terrorism. Accomplishments include the following:
- co-developed the Blood Availability and Safety Inventory System (BASIS) used by DHHS to monitor the availability of blood components at major hospital centers and blood establishments throughout the U.S.
- established the CBER Blood Safety Team to improve CBER's response to blood safety issues, and to enhance external outreach, evaluation, and risk communications
- facilitated continual development of new medical countermeasures (e.g., rPA vaccines, anthrax immune globulin, new smallpox vaccines, and botulinum antitoxin) through expedited regulatory review and assistance to manufacturers
- completed the Final Order on the Efficacy Review of Bacterial Vaccines and Toxoids, including the anthrax vaccine—this review was published in the Federal Register on December 19, 2005 (Biological Products, Bacterial Vaccines and Toxoids, Implementation of Efficacy Review, Anthrax Vaccine Adsorbed, Final Order)
- reassessed and strengthened response capabilities including the development of continuity of operations plans
- proactively identified gaps that exist related to needed medical countermeasures against biological agents that could be used in an attack
lead effort to finalize new labeling regulations for medical products purchased for the Strategic National Stockpile - studied/developed improved immunological assays for anthrax vaccines, the protective isotypes of vaccinia immune globulin, correlates of immunity for tularemia, the cellular trafficking of botulinum toxin, and stimulation of innate immunity against various agents
- held numerous pre-IND/technical meetings with potential manufacturers of medical countermeasures to assist in their development and the submission of INDs, as well as development of their licensure path.
*See Appendix A References 30-38
Enhance Patient And Consumer Protection And Empower Them With Better Information About Regulated Products
CBER actively addresses the highest public health priorities through collaborative, innovative and cooperative actions. The Center has emphasized the importance of outreach, new paths to product evaluation, increased access to products, and safety. Improving blood safety through new blood donor screening tests and safety surveillance of licensed biological products to detect new information about adverse events have significantly increased public health protections. Increasing the amount of information available to guide consumer decisions on product use and the elimination of false and misleading product information contribute to a public better informed about biologic products.
Improving the Safety of the Blood Supply with Novel Donor Screening Tests*
Improvements in blood donor screening and testing over the last few years have helped make the nation's blood supply safer from infectious diseases than it has been at any other time in history. A number of products and product supplements have been approved in the last fiscal year that contribute to this effort.
Hepatitis B
Hepatitis B (HBV) is caused by a virus that infects the liver. The virus, which is transmitted by blood, can, in some cases, cause lifelong infection, cirrhosis (scarring) of the liver, liver cancer, liver failure, and death. This past year, CBER approved the first fully automated tests for hepatitis B virus (the Abbott Prism HBcore assay, the Abbott Prism HBsAg assay, and the Abbott Prism HBsAg Confirmatory assay). The Abbott Prism automated test system increases the efficiency and convenience of screening blood, tissue, and organ donors for HBV. The Abbott Prism tests are fully automated, thus reducing the potential for operator errors. They are tamper-resistant, with redundant checks to ensure integrity of the testing system, and highly sensitive and specific for detection of infection with HBV.
West Nile Virus
WNV is typically transmitted to humans by mosquito bites. It was first detected in the U.S. in 1999, and the virus has recurred each year for seven consecutive years, causing close to 20,000 human cases of disease and at least 762 deaths since 2002. It has been estimated between 1 and 2 million people have been infected with WNV. In 2002, it was discovered the WNV could be transmitted in blood, and an urgent effort to develop a blood test began. With support from FDA, CDC, and NIH, manufacturers developed investigational WNV NATs that were rapidly put in place to evaluate their effectiveness and (as an interim measure) to protect the blood supply. Blood banks across the U.S. participated in these efforts, to safeguard the blood supply.
CBER approved the first WNV blood test to screen donors of blood, organs, cells, and tissues in December 2005. The Procleix WNV assay, developed by Gen-Probe, Inc. and marketed by Chiron Corporation, detects viral genetic material (ribonucleic acid or RNA). This new test will help protect patients who receive blood and other biologic products against WNV infection. This approval is the result of a tremendous cooperative effort among FDA, other public health agencies, state health departments, test kit manufacturers, and the blood industry. With the approval of a screening test for WNV, CBER is developing guidance for industry on the implementation of these tests.
Despite all the progress in testing and improvement in the understanding of WNV, scientific questions still remain. CBER is also conducting studies on the partitioning of WNV in blood components to determine whether new approaches to testing, including the use of whole blood as the test sample, could increase the sensitivity of WNV NAT, thereby further reducing the risk of WNV from NAT screened donations.
*See Appendix A References 39-40
Monitoring the Availability of Blood and Blood Components*
The TRANS-Net Blood and Reagent Shortage Monitoring System, designed and piloted by CBER, has been incorporated into the DHHS Public Health Emergency Management System. Blood and blood components are critical biological products that provide life support to patients in the U.S. each year and are frequently needed in large quantities during times of natural disaster and other emergencies. More than 29 million units of whole blood, red blood cells, and platelets are transfused each year. However, these products have relatively limited storage times and are critically dependent on the routine availability of eligible donors, as well as supplies for collection of blood and reagents for testing.
Systems for assessing national blood supply inventories, while successfully measuring historical trends in collection and utilization, have not previously had the capability to assess local, regional, and national blood component shortages in real time. In response to the September 2001 tragedy, TRANS-Net was designed at CBER as a Web-based system that provides a mechanism for voluntary real-time reporting of shortage information to a central site from the nation's 1,848 registered blood collection sites and 2,348 transfusion services. These reports are received electronically and mapped at a central data monitoring facility. Data are then assessed manually and electronically through the use of software that recognizes the geospatial density and frequency of information received. This software creates an alert message when a predetermined shortage threshold is reached. It also creates geospatial maps reflecting shortage levels that can be integrated with other mapped data (e.g., hospital census) that are also part of the BASIS blood inventory monitoring system, which is housed in the DHHS Secretary's Operations Center. Incoming data also contain information that helps to indicate the seriousness of a reported shortage event, for example:
- delayed surgery or medical procedure (number of events per reporting day)
- support of Rh-negative patients with Rh-positive blood (number of events per reporting day)
- unfilled (or underfilled) blood orders (e.g., <50% of requested blood received)
- triage of available supply
- insufficient blood to meet emergency needs.
As part of the TRANS-Net design, summary reports of appropriate blood availability information will be made available on the Web to public health stakeholders and eventually to the general public as an aid in recruiting blood donors.
Enhancing the Safety of Blood and Development of the CBER Blood Safety Team
The CBER Blood Safety Team was launched in July 2006. Led by the Office of Blood Research and Review (OBRR), the interdisciplinary Blood Safety Team is comprised of experts from several CBER offices, including the Office of Compliance and Biologics Quality (OCBQ), the Office of Communications, Training, and Manufacturers Assistance (OCTMA), Office of Biostatistics and Epidemiology (OBE), and the Immediate Office of the Director (IOD). Four main goals of the Blood Safety Team are to:
- improve CBER's response to blood safety issues
- improve the availability and quality of CBER's blood safety information
- improve processing of blood safety information
- enhance CBER's external outreach, evaluation, and risk communication.
Current objectives of the Blood Safety Team include the following four areas:
- finalizing the blood safety reporting rules and guidances
- evaluating methods to improve the collection and assessment of adverse events, transfusion related fatalities, and Biological Product Deviation Reports (BPDRs)
- developing pathways to track plasma derivatives
- increasing access and partnerships with other DHHS agencies.
We anticipate the activities of the Blood Safety Team will enhance CBER's ability to monitor and respond to blood safety issues and further protect the nation's blood supply and blood products from infectious diseases and other emerging threats.
Biological Safety Activities*
A fundamental aim in CBER's continuous safety surveillance of licensed biological products is to enhance our understanding of the safety profile of the products we regulate and to provide updated safety information to the public. Our efforts can help physicians weigh a product's risks and benefits and help patients to use these products as safely as possible. CBER also presents information to advisory committees and publishes articles in the medical literature about the safety of biological therapeutic and prophylactic products.
CBER has played an important new role in implementing the International Conference on Harmonisation's (ICH) Pharmacovigilance Planning Guidance. CBER recognizes the importance of pharmacovigilance plans submitted as part of the license application, and our review of such plans is an important activity in the area of postmarketing safety.
*See Appendix A References 42-44
CBER Outreach Update
CBER's outreach efforts during FY 2006 focused on improving the Center's communication through innovative communication strategies. CBER's outreach program reaches tens of thousands of stakeholders annually, both directly and indirectly, including consumers, health care professionals, regulated industry, members of Congress, and the media.
During FY 2006, CBER began an initiative to improve the organization and quality of information on its website. The website serves as a focal point for obtaining information from the Center. The redesign will make information easier to find. There were almost 18 million hits to the site during FY 2006, averaging approximately 43,000 hits per day. The Center also maintains three automated e-mail distribution lists, which now reach more than 8,000 subscribers. These "listservs" allow CBER to proactively distribute information electronically, reaching a wide audience quickly and efficiently.
Over the past several years, CBER has enjoyed a successful exhibit program, and FY 2006 was no exception. The Center participated in annual meetings, conferences, and workshops. The Center's exhibit program reaches a vast array of constituents, including those working in regulated industry, counterterrorism research, infectious diseases and infection control, and parents' groups (see Appendix B).
CBER held several workshops in FY 2006 that were intended to provide important information to researchers, product sponsors, and the public on key topics related to product development and licensure. CBER also participated in information-sharing liaison meetings with trade associations and developed several co-sponsorship agreements for workshops as avenues to disseminate important regulatory and policy information. In addition, CBER contributed a number of important updates on product approvals and safety information to FDA Patient Safety News, a monthly broadcast produced by the Agency and delivered to hospital networks around the country.
Increase Access To Innovative Products And Technologies To Improve Health
CBER continues to meet or exceed the fee performance goals for drugs and devices, thereby increasing the number of biological products and related devices available to those in need
. Newly approved products, such as RotaTeq to prevent rotavirus gastroenteritis in infants, Gardasil to prevent cervical cancer caused by certain human papillomaviruses (HPV), and Zostavax to reduce the risk of shingles all address important public health needs. The Critical Path initiative focuses on facilitating innovative products for the 21st Century. The Center is working to advance the development of more efficient vaccine production which will benefit the public health in many areas, including strengthening our capacity to deal with seasonal and pandemic influenza.
Initiatives to Strengthen, Diversify, and Increase Capacity for Influenza Vaccines
The 2005-2006 Influenza Season
Seasonal influenza is a significant cause of morbidity and mortality in the U.S. According to CDC, every year on average in the U.S., approximately 5 percent to 20 percent of the population get sick with influenza, more than 200,000 people are hospitalized from its complications, and approximately 36,000 people die from it. Some individuals, such as the elderly, children, and people with certain chronic medical conditions, are at high risk for serious complications from influenza. Ninety percent of influenza-related deaths occur in individuals aged 65 or older; however healthy children under 2 years of age are as likely to be hospitalized because of influenza as older adults.
Influenza vaccine is unique because its active ingredients, the virus strains used to manufacture the vaccine, change almost every year. Because the circulating viruses are constantly mutating, each year's vaccine usually differs from the preceding year. Therefore, manufacturers must produce tens of millions of doses of a new vaccine each year. Promising technologies (e.g., cell culture and recombinant protein and DNA-based influenza vaccines) are in the research and development stages. In September 2006, CBER issued draft guidance on development of cell culture vaccines that is applicable to the development of influenza vaccine. CBER is working with our DHHS colleagues and with manufacturers to advance their development. The vaccine manufacturing process is complex and can create uncertainty for the vaccine supply. Preparing for the influenza season each year is a time-critical, collaborative effort involving FDA, CDC, NIH, WHO, vaccine manufacturers, and the health care community.
The strain selection process usually occurs in February when an FDA advisory committee meets to recommend which three strains of the virus should be included in the vaccine, based on data from WHO laboratories in more than 80 countries. FDA makes the final decision on which strains will be included in the vaccine for the
U.S. population.
FDA, CDC, other WHO collaborating centers, and other partners produce reference influenza viruses that represent the selected strains and are adapted to high growth in eggs. The reference influenza viruses are provided to the licensed vaccine manufacturers to generate the "seed virus" for further manufacturing the influenza vaccine. Manufacturers then inject the seed viruses into fertilized chicken eggs, and the virus multiplies.
CBER produces and provides manufacturers with antiserum that is used to test vaccine potency for each influenza strain used in the manufacture of trivalent inactivated influenza vaccines. Manufacturers of both inactivated and live attenuated influenza vaccines ship samples of vaccine from each lot, along with their test results, to CBER for lot release. CBER reviews the manufacturer's test results and also may perform its own tests on the vaccine before releasing each lot for distribution.
It takes about 6 months to complete influenza vaccine production, from egg to vial, each season. FDA interacts extensively with licensed manufacturers to address issues that may arise during annual production and to facilitate resolution of these issues. In addition, since 2004, FDA has conducted inspections of influenza vaccine manufacturers on an annual basis to identify problems earlier and prevent them whenever possible. Although some lots of vaccine may be released as early as July, manufacturing usually continues until October or later to produce and test the large volume of vaccine required for the U.S. population.
In August 2005, FDA announced the approval of Fluarix, an influenza vaccine for adults that contains inactivated virus. Fluarix is approved to immunize adults 18 years of age or older against influenza virus subtypes A and type B contained in the vaccine. The approval of Fluarix broke new ground in that it was the first vaccine approved using FDA's accelerated approval process. Accelerated approval allows FDA to approve products for serious or life-threatening diseases based on early evidence of a product's effectiveness; therefore reducing the time it takes for needed medical products to become available to the public. In this case, the manufacturer demonstrated that, after vaccination with Fluarix, adults made levels of protective antibodies in the blood that FDA believes are likely to be effective in preventing influenza. GlaxoSmithKline, manufacturer of Fluarix, is performing further clinical studies as part of the accelerated approval process to verify the clinical benefit of the vaccine. In the fall of 2006 and in time for the 2006-07 influenza season, FDA approved ID Biomedical's trivalent inactivated influenza vaccine, FluLaval, to immunize adults 18 years of age and older against influenza disease caused by virus subtypes A and type B. FDA again used the accelerated approval pathway.
Four manufacturers distributed influenza vaccine for the 2005-2006 season:
- sanofi pasteur;
- MedImmune Vaccines, Inc.;
- GlaxoSmithKline Biologicals; and
- Chiron
The estimated supply of vaccine for the 2005-2006 season was 86 million doses, approximately 40 percent more than the 2004-2005 season's 61 million doses. Demand for vaccine averages 70 million to 75 million doses annually.
The peak demand for influenza vaccine is in October and November. Because the influenza disease season usually peaks in January or later in the U.S., FDA and CDC support extending vaccination into January and February of each year.
FDA Issues Advice to Facilitate Influenza Vaccine Development
In March 2006, CBER's Office of Vaccines Research and Review (OVRR) released two draft guidance documents, one for seasonal influenza vaccines (annual trivalent inactivated) and the other for pandemic influenza vaccines, to aid manufacturers in evaluating seasonal and pandemic influenza vaccines for safety and effectiveness. FDA's goal is to increase the supply of safe and effective influenza vaccines by outlining the regulatory pathway for the rapid development and approval of influenza vaccines. The two guidances provide manufacturers with guidelines on the clinical data needed to show safety and effectiveness for new influenza vaccines. Consistent with the goals of FDA's Critical Path Initiative to get products to market more quickly and to advance the development and use of new technologies, these documents outline specific approaches for vaccine developers to follow. In essence, through the guidances, FDA provides a road map to assist industry and provides advice for new manufacturers on how to develop new and safe vaccine products more quickly.
Additional and more specific information on the guidances can be found on the CBER website at www.fda.gov/cber/guidelines.htm.
Technology for New-Generation Vaccines for Viral Infectious Diseases
In an effort to aid manufacturers in developing safe and effective cell-based viral vaccines, including those to address emerging and pandemic threats, FDA issued a new draft guidance in September 2006. "The guidance document…is a vital part of our overall efforts to help manufacturers develop new vaccines that are critical to meeting global public health needs," said then acting FDA Commissioner, Andrew von Eschenbach, M.D.
"This guidance promises to help modernize the development of life-saving vaccines for influenza and other diseases and facilitate the development of more plentiful, reliable supplies."
In the guidance, FDA provides manufacturers of viral vaccines with updated recommendations to the 1993 document, "Points to Consider in the Characterization of Cell Lines Used to Produce Biologics." The guidance document conveys information for determining the suitability of a cell culture for manufacturing, which includes testing and validating the safety and purity of the cells used in the development and production of viral vaccines.
While cell cultures currently are used in the manufacture of many licensed vaccines that help protect against diseases, such as polio, rubella, and chickenpox, the new guidance further helps facilitate the development of additional vaccines. The guidance outlines the best practices using current and emerging science. Cell-based vaccine manufacturing holds the promise of a reliable and flexible alternate method of producing influenza vaccines, which are currently produced in chicken eggs by a technique developed more than 50 years ago. With increasing demand for seasonal influenza vaccine and with the potential threat of a pandemic, as well as other emerging infectious diseases (e.g., SARS), more flexible approaches that allow surge capacity in an emergency are critical. With cell-based manufacturing, cells can be frozen, stored, and thawed as needed to produce more vaccine.
A copy of the draft guidance, "Guidance for Industry: Characterization and Qualification of Cell Substrates and Other Biological Starting Materials Used in the Production of Viral Vaccines for the Prevention and Treatment of Infectious Diseases," is available at www.fda.gov/cber/gdlns/vaccsubstrates.pdf.
Critical Path Initiative: Personalized Medicine*
Critical Path is FDA's premier initiative to identify and prioritize the most pressing medical product development problems and the greatest opportunities for rapid improvement in public health benefits. Its primary purpose is to ensure basic scientific discoveries translate more rapidly into new and important medical treatments by creating new scientific tools to develop and evaluate new medical products.
On March 16, 2004, FDA released a report, Innovation/ Stagnation: Challenge and Opportunity on the Critical Path to New Medical Products, addressing the apparent slowdown in innovative medical therapies submitted to FDA for approval. The report describes the urgent need to modernize the medical product development and evaluation process (the Critical Path) to make product development more predictable and efficient. Because of its unique vantage point, FDA can work with companies, patient groups, academic researchers, and other stakeholders to coordinate, develop, and/or disseminate solutions to scientific hurdles that are impairing the efficiency of product development industry-wide.
Through the Critical Path Initiative, FDA took the lead in the development of a Critical Path Opportunities List and Report. The report describes and provides examples of how new scientific discoveries in fields, such as genomics and proteomics, imaging, and bioinformatics, can be used in bringing products to market, and provide the right treatment at the right time to the right patient. The Opportunities List was released on March 16, 2006, by Secretary Leavitt and Commissioner von Eschenbach (then Acting Commissioner). The Opportunities List outlines an initial 76 projects to bridge the gap between the quick pace of new biomedical discoveries and the slower pace with which those discoveries are currently developed into therapies. The release of the Opportunities List marks a starting point in identifying priorities to be accomplished under the Critical Path Initiative. Government, industry, and academic experts estimate that, if accomplished, the new tests and tools developed under the Critical Path Initiative will modernize the drug development process by 2010 and help make treatments safe while bringing new medical discoveries to patients faster and at a lower cost. The Opportunities List is available at http://www.fda.gov/oc/initiatives/criticalpath/reports/opp_report.pdf.
CBER continues to leverage resources with other government agencies. In January 2006, FDA, the National Cancer Institute (NCI), and Centers for Medicare and Medicaid Services entered into a Memorandum of Understanding for the Biomarker Qualification Initiative. The three agencies collaborate and pursue specific projects aimed at improving the clinical utility of biomarkers as diagnostic and assessment tools that facilitate the development of safe and more effective cancer therapies.
CBER has a long tradition of research aimed at resolving challenges to biological product evaluation. Many of these are cited within the Annual Report text. Additional recent Critical Path achievements of CBER can be viewed at www.fda.gov/oc/initiatives/critical path/opportunities06.html.
Research Management Advances
FY 2006 marked a year of major milestones in CBER research management and successes. CBER research and scientific expertise contribute to facilitating product development and evaluation. CBER has expert medical, scientific, and technical staff who are members of interdisciplinary review teams. Some of these individuals also perform research to advance biological product safety, efficacy, and quality.
CBER uses the following principles to facilitate the management of research programs and objectives within the Center:
- The CBER research program is highly collaborative and includes laboratory, epidemiological, statistical, and clinical sciences.
- Its scope encompasses the scientific basis of preclinical and clinical studies, manufacturing, regulatory submissions, inspections, postmarketing surveillance, and stakeholder outreach (e.g., guidances).
- The research is high-quality, efficient, and provides outcomes that address scientific and regulatory challenges in product development, safety, efficacy, and quality.
This past year, CBER established a Research Leadership Council (RLC) to spearhead the CBER Research Management Initiative. The RLC developed and is implementing a transparent, mission driven research program management process. CBER is developing a center-wide Scientific Research Plan, for which input from CBER's Advisory Committees will be sought.
Scientific Expertise Teams
CBER has a wide range of scientific expertise. The RLC developed the Scientific Expertise Team Matrix to identify and manage the scientific and technical expertise within the Center. This matrix provides a mechanism to best utilize CBER scientific resources for product regulation by identifying specific scientific expertise within CBER.
CBER regularly invites external scientific experts on advisory committees to evaluate, through site visits, the achievements and future plans of the CBER Research Laboratories and research-regulator staffs. CBER expanded external review of research to include a broader programmatic assessment of each product office's research program (OVRR, OBRR, and the Office of Cell, Tissue, and Gene Therapy (OCTGT)). These evaluations are important tools in our annual research planning.
Scientific Training Initiatives
CBER also has a long tradition of collaborative research. In FY 2006, we developed a program to enhance research training of collaborative scientists. Through this program collaborative research opportunities are established with other domestic and international governmental and nongovernmental institutions. The Collaborative Scientific Training Program (CSTP) provides an opportunity for acquiring additional scientific expertise within CBER, as well as training a cadre of researchers (e.g., postdoctoral students) in the science of product development and regulatory research. Inaugural CSTP partners include the Korean FDA and the University of Yokohama in Japan, and further outreach is underway with other domestic and international academic and regulatory institutions.
CBER medical, scientific, and technical staffs use their expertise to evaluate complex biological medical products. It is critical to continually update and advance their expertise in concert with the rapid advancements in 21st century science. In FY 2006, CBER developed a program to support continuing technical, scientific, and medical education initiatives for scientific staff. Through this program, these staff were able to attend seminars and educational meetings in their area of medical, scientific, and technical expertise.
To provide formal education in the science of leadership and management, CBER developed and offered a series of leadership and management training courses for all personnel. The courses included Productive Conflict Management, Situational Leadership, and Motivating Your People. These courses were well-attended and are an essential component in staff development and succession planning at CBER.
Selected FY 2006 research accomplishments at CBER include the following:
- established programs to develop and evaluate enhanced approaches to pandemic influenza vaccine manufacture, safety, and efficacy
- identified biomarkers that may serve as indicators of epithelial ovarian cancer disease progression/ response to therapy; identified portion of Ebola virus critical to cell entry, which may provide a target for vaccine development
- embarked on international study with the WHO to qualify a new, small animal mumps vaccine safety test
- developed a new assay to distinguish HIV-vaccinated subjects from HIV-infected subjects in clinical trials
- commenced a new proteomics coordination effort to enhance and modernize biological product characterization techniques, including influenza vaccine, cell substrates, and gene therapies.
Selected FY 2006 partnering relationships include the following:
- studies relating to cell substrate testing for vaccine manufacture, HIV-related biological product evaluation and development
- Interagency Oncology Taskforce fellowship program with the National Cancer Institute (NCI)
- support of CBER core biotechnology services with the National Institute of Dental and Craniofacial Research (NIDCR) and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)
- ongoing development of small animal models for preclinical testing of tularemia vaccines, aerosolized anthrax, smallpox vaccine, and SARS vaccine
- preclinical assay development for tuberculosis vaccines with the Aeras Global TB Vaccine Foundation
- proteomics for blood product characterization with the Alpha-1-Foundation
- transgenic mouse model development with the American Red Cross to address variant Creutzfeldt-Jakob disease (vCJD) contamination
- genomics analysis of cell substrate quality with the American Type Culture Collection
- development of DNA microarray system for detection of pathogenic agent contamination of biological products with Johns Hopkins University
- improvement of safety and production of pandemic flu vaccines with the National Vaccine Program Office, including improving cross-protection of influenza vaccines.
*See Appendix A References 45-51
User Fee Programs
PDUFA
In 1992, Congress passed the Prescription Drug User Fee Act (PDUFA). Congress reauthorized PDUFA in the FDA Modernization Act of 1997 (FDAMA), and again in the Public Health Security and Bioterrorism Preparedness and Response Act of 2004. The PDUFA authorizes FDA to collect fees from companies that produce certain human drug and biological products. When a company seeks FDA approval for a new drug or biologic prior to marketing, the company must submit an application along with a fee to support the review process. In addition, companies pay annual fees for each manufacturing establishment and for each prescription drug product marketed. In this program, industry provides added resources needed to meet review performance goals, which emphasize timeliness while maintaining safety and efficacy of FDA-approved products.
PDUFA has provided FDA with needed resources for the review of human drug and biologic applications. Fees are used to help reduce the time required for evaluating human drug applications and to support review quality. FDA submits annual performance and financial reports to Congress on progress in streamlining the drug review process and use of PDUFA fees.
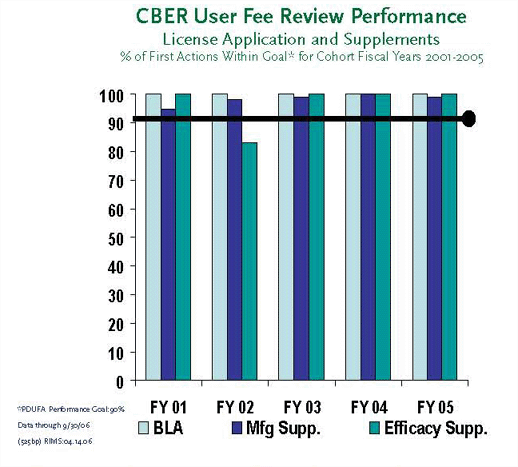
On November 10, 2005, FDA also issued a white paper entitled, "Adding Resources and Improving Performance in FDA Review of New Drug Applications." The document shows the evolution of the PDUFA program since its enactment in 1992 and progress to date. The paper also discusses how fees are collected and how the fees are utilized.
On November 14, 2005, FDA held a public meeting entitled, "Prescription Drug User Fee Act: Public Meeting." This meeting included presentations by FDA and a series of panels representing different stakeholder interest groups (e.g., patient advocates, consumers, industry members, health professionals, and academic researchers). The public meetings shared stakeholders views as FDA prepared to work on amended authorizing legislation for PDUFA.
The authority for PDUFA III expires in September 2007; without further legislation, FDA would no longer be able to collect user fees for the prescription drug program, and resources critical to running the program would become unavailable to FDA. Information on FDA's PDUFA performance and financial reports, including the white paper, can be accessed at www.fda.gov/oc/pdufa.
MDUFMA
The Medical Device User Fee and Modernization Act of 2002 (MDUFMA) is providing needed funds to FDA for "the review of devices and the assurance of device safety and effectiveness so that statutorily mandated deadlines may be met." MDUFMA user fees coupled with the additional appropriations required by MDUFMA, have helped reverse that trend and are providing the following continuing benefits:
- Safe and effective devices used to diagnose and treat disease are reaching the public more rapidly.
- Manufacturers are receiving timely, high-quality application reviews.
Devices marketed in the U.S. continue to meet high standards for safety and effectiveness.
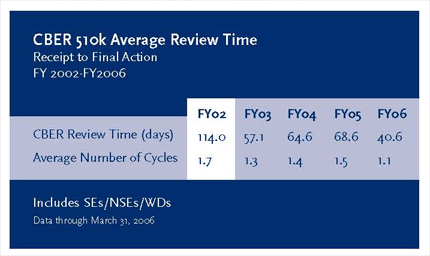
CBER interaction with government partners and industry has facilitated the recent approval of rapid tests for HIV and tests to monitor HIV drug resistance, examples of successful regulation under the framework established by MDUFMA. The Center aims to apply regulation in a risk-based manner. Certain areas in CBER's oversight, including blood-screening tests, raise unique concerns. CBER seeks to address these in a balanced, transparent, and least burdensome manner, and welcomes public and industry input as we develop guidance to industry on devices regulated by CBER. On April 28, 2006, CBER, in conjunction with CDRH, issued guidance to industry that provided information about the real-time review process for premarket approval application (PMA) supplements and outlined the procedures for requesting and submitting these types of documents. On August 1, 2006, CBER/CDRH also issued guidance to industry entitled, "Small Business Qualification Worksheet and Certification," that provided updated information regarding which firms qualify for small business fees and discounts for FY 2007.
On September 30, 2007, the user fee provisions of MDUFMA will expire. To help FDA and all stakeholders evaluate the program and prepare for possible new legislation to reauthorize MDUFMA, FDA sought input from interested parties about those aspects of MDUFMA that worked well and those areas for which change should be considered. FDA held its Third Annual MDUFMA Stakeholders meeting on November 17, 2005. The meeting provided a forum for industry, the general public, and all stakeholders to give feedback to FDA on how the user fee process is working and recommendations for improvement of the device review program.
Negotiations with industry for MDUFMA II include representatives from CBER, CDRH, Office of Regulatory Affairs (ORA), and the Office of the Commissioner. CBER has met or exceeded the MDUFMA review performance goals for FY 2005. FDA's MDUFMA performance and financial reports can be accessed at www.fda.gov/oc/mdufma.
Cellular and Gene Therapies: Facilitating Availability and Development of Safe and Effective Products Through New Technologies
Genomics and Proteomics
As hundreds and thousands of endpoints are analyzed simultaneously, genomics and proteomics technologies offer novel approaches to understanding biological processes. Not only are these technologies being incorporated into the routine of academic laboratories, but they are also becoming a tool for biotechnology, product characterization, and clinical research conducted by academia, government agencies, and industry. The results of genomic and proteomic studies have begun appearing in INDs and BLAs/New Drug Applications (NDAs) submitted to FDA. To facilitate development and availability of safe and effective technologies, CBER is engaged in the research and development of standards, performing critical path research to characterize cell substrates and products, including cellular and gene therapy products and developing guidance documents in the area of genomics and proteomics technologies. CBER is also engaged in developing expertise by providing a hands-on training program for regulatory scientists and research reviewers. More than 50 regulatory scientists/research reviewers have been trained through this program.
CBER helped develop FDA guidance for industry, "Pharmacogenomic Data Submissions," released in March 2005. CBER scientists are also involved in FDA's Interdisciplinary Pharmacogenomics Review Group (IPRG), which evaluates voluntary genomics data that sponsors submit to FDA. To keep pace as science and technology evolves, in the past year an expert working group on "Pharmacogenomics" was formed as part of ICH. Both CBER and CDER staff, along with representatives from Europe and Japan, participate in this group. The primary objective of this ICH group is to define basic terminology pertaining to pharmacogenomics, pharmacogenetics, and methods for coding of pharmacogenomic samples and data. Other objectives are to define terminology of genomic biomarker attributes. This group has begun drafting a guideline that will harmonize pharmacogenomic terminology and define consistent terms for use in regulatory documentation. This guideline will help facilitate integration of this evolving science into global drug development.
Tissue Engineering
In support of the critical path element entitled, "Tools for Assessing Safety, Demonstrating Medical Utility, and Characterization of Manufacturing," CBER, together with CDRH, is developing a partnership with other federal agencies to advance tissue-engineering science and to facilitate the development of safe and effective tissue-engineered products. The CBER/CDRH Tissue Engineering Steering Committee promotes inter-center interactions on topics of mutual interest. CBER staff also participate on the CDRH Specialty Task Group on Tissue Engineering, which is responsible for coordinating Agency interactions with the American Society for Testing and Materials International (ASTM) in the development of their international standards for Tissue-Engineered Medical Products (TEMP). CBER and CDRH staff attended the inaugural Tissue Engineering/ Regenerative Medicine (TERM) liaison meeting on April 28, 2006. Topics included FDA regulatory review process specific to cell scaffold products, key clinical challenges, unique product testing paradigms, and manufacturing issues in development of TERM products.
CBER staff participate in the Multi-Agency Tissue Engineering Science (MATES) Interagency Working Group, organized under the auspices of the Subcommittee on Biotechnology, National Science and Technology Council (NSTC). The primary goal of MATES is to facilitate communication across departments/ agencies by regular information exchanges and to enhance cooperation through co-sponsorship of scientific meetings and workshops. This past year, members of the MATES Working Group have been preparing a document entitled, "Strategies for Advancing Tissue Science and Engineering: Foundation for the Future." This document, when completed, will provide a strategic plan for the advancement of tissue engineering in the federal government. CBER has met with product manufacturers currently developing these innovative products and has a unique perspective pertaining to the issues associated with the early phases of developing tissue-engineered products. The need to provide a clear strategy that defines the types of studies and data essential for supporting regulatory submissions has been consistently noted. For example, CBER has identified a need for criteria suitable for the characterization of final manufactured cell-scaffold, tissue-engineered products. Key issues being considered include the following:
- What questions should be asked and addressed by testing, and at what stage of product assembly?
- What testing methods are available and what methods need to be developed?
Cellular and Gene Therapies: Outreach and Partnerships
CBER has provided proactive scientific and regulatory guidance in areas of novel product development. The Center communicates regulatory expectations and encourages dialogue on cutting-edge product development to help define the best scientific approaches and reduce product development time and risk. Early and continuing interactions with stakeholders and the public have proven to be an effective means of communicating, and addressing issues regarding potential risks and benefits, thus avoiding unnecessary regulatory burdens.
Following are some examples of these interactions during the past fiscal year:
- On January 27, 2006 and June 16, 2006, CBER staff attended the Cell Therapy/FDA Liaison Meetings. Topics discussed included HIV and HCV NAT testing for donor of human cells and tissues, and microbial contamination of peripheral blood progenitor cell products.
- In February 2006, CBER staff held a Cellular, Tissues, and Gene Therapies Advisory Committee meeting to discuss the complex and difficult area of potency assay development for cell and gene therapy products. CBER staff discussed a matrix approach to potency assays to spur development of complex products. The meeting also included a session on the NTP Proposed Study on Retroviral Vector-Mediated Insertional Mutagenesis.
As part of the NTP, and in collaboration with the National Institute of Environmental Health Sciences (NIEHS), CBER initiated studies to develop approaches to reduce the risk of cancer following retroviral gene therapy. Included in this collaboration are preclinical studies involving DNA-based therapeutics, specifically focusing on retroviral vector safety aimed at reducing the risk of retroviral-mediated tumorigenesis following administration into humans. The goal of the project is to reduce the uncertainty in the risk assessment/risk benefit analysis; enable a better estimate of risk; and establish high-quality, science-based safety assessments/risk management decisions for the consumer.
As part of our outreach efforts, CBER staff participated in numerous national, international, trade, and industry meetings. CBER has also been involved with liaison meetings within FDA involving Alzheimer's disease and the use of cellular therapy in cardiovascular disease. Much of our effort has been directed toward specific disease processes or emerging technologies. CBER staff has also worked extensively with the NIH extramural program on trials to establish the safety and efficacy of pancreatic islet cell transplantation for the treatment of diabetes mellitus.
To further communicate our regulatory expectations, CBER has issued the following guidances: "Chronic Cutaneous Ulcer and Burn Wounds—Developing Products for Treatment" (with CDRH) and "Guidance for Industry: Gene Therapy Clinical Trials— Observing Participants for Delayed Adverse Events." On July 27, 2006, CBER staff presented a talk entitled, "National Policies for Xenotransplantation Studies in the USA." In addition, CBER represented DHHS at a WHO consultation on September 27-29, 2006, entitled, "Second Global Consultation on Critical Issues in Human Transplantation," in which international policies for xenotransplantation oversight were discussed.
FDA Approves Gardasil
On June 8, 2006, FDA approved Gardasil, the first vaccine developed by Merck & Co., Inc. to prevent cervical cancer; precancerous genital lesions; and genital warts due to human papillomavirus (HPV) types 6, 11, 16, and 18. The vaccine is approved for use in females 9-26 years of age. HPV types 16 and 18 account for approximately 70 percent of cases of cervical cancer, whereas HPV types 6 and 11 cause approximately 90 percent of genital wart cases. HPV is the most common sexually transmitted infection in the U.S. CDC estimates that about 6.2 million Americans become infected with genital HPV each year and that more than half of all sexually active men and women become infected at some time in their lives. Most infected women will clear the virus and do not develop related health problems. However, some HPV types can cause abnormal cells on the lining of the cervix that can turn into cancer years after infection with the virus. In the U.S., on average, there are 9,710 new cases of cervical cancer and 3,700 deaths attributed to it each year. Worldwide, cervical cancer is the second most common cancer in women and is estimated to cause more than 470,000 new cases and 233,000 deaths each year.
The license application for Gardasil was evaluated and approved in 6 months under FDA's priority review process, which is targeted to products that provide significant health benefits over existing products. Gardasil is a recombinant protein vaccine (genetically engineered) that is given as three injections over a 6-month period. In women who were not already infected with HPV, Gardasil was nearly 100 percent effective in preventing precancerous cervical lesions, precancerous vaginal and vulvar lesions, and genital warts caused by the HPV types against which the vaccine is directed. Immunization with Gardasil also is expected to prevent most cases of cervical cancer due to HPV types targeted by the vaccine. "This is the first vaccine licensed specifically to prevent cervical cancer. Its rapid approval underscores FDA's commitment to help make safe and effective vaccines available as quickly as possible. Not only have vaccines dramatically reduced the toll of diseases in infants and children, like polio and measles, but they are playing an increasing role protecting and improving the lives of adolescents and adults," said Jesse Goodman, M.D., M.P.H., Director of CBER.
FDA Approves Zostavax
On May 25, 2006, FDA approved Zostavax manufactured by Merck & Co., Inc., which is the only vaccine indicated to prevent shingles (herpes zoster) in people 60 years of age and older. Zostavax, a live virus vaccine, boosted immunity against varicella-zoster virus, the same virus that causes chickenpox. The studies showed, overall, in those ages 60 and above, the vaccine reduced the occurrence of shingles by about 50 percent. The vaccine is given as a single injection under the skin, preferably in the upper arm.
After an attack of chickenpox, the varicella-zoster virus lies dormant in certain nerve tissue but can reactivate and appear as shingles years later. Shingles can occur in people of all ages, but most commonly in those greater than 60 years of age. The risk of singles increases as people get older, and shingles is estimated to affect 2 in 10 people in their lifetime. When shingles develops, a rash or blisters appear on the skin, generally on one side of the body. This is a sign the virus, that has been dormant in the nerve cells, has reactivated and traveled from the nerves and followed a path out to the skin. Because the nerves along the path become inflamed, shingles can also be painful. Pain that lasts for months after the rash has healed is called postherpetic neuralgia. For some people, this pain can be severe and chronic. "This vaccine gives health care providers an important tool that can help prevent an illness that affects many older Americans and often results in significant chronic pain," said Jesse L. Goodman, M.D.,
M.P.H.
FDA Approves RotaTeq
On February 3, 2006, FDA announced the approval of RotaTeq manufactured by Merck & Co., Inc. for preventing rotavirus gastroenteritis in infants between the ages of 6 and 32 weeks. RotaTeq is a live-virus vaccine that is given by mouth in three doses. Infection with rotavirus is a leading cause of severe diarrhea in infants and young children in the U.S. and worldwide and may also cause vomiting, fever, and dehydration. CDC has estimated that rotavirus infection results in approximately 55,000 hospitalizations annually of infants and young children in this country. Death from rotavirus is rare in the U.S. However, in developing countries, rotavirus gastroenteritis has been estimated to cause several hundred thousand deaths annually in infants and young children. "This vaccine gives health care providers an important new tool that can effectively prevent an illness that affects almost all children within the first few years of life," said Jesse L. Goodman, M.D., M.P.H.
Overall, approximately 72,000 healthy infants were studied in the United States and other countries in randomized placebo-controlled studies to look at the safety of RotaTeq. Of these infants, almost 7,000 from the U.S. and Finland were also studied for efficacy. In these studies, RotaTeq prevented 74 percent of all rotavirus gastroenteritis cases and 98 percent of the severe cases. In addition, RotaTeq prevented approximately 96 percent of hospitalizations due to rotavirus gastroenteritis.
In 1998, FDA approved a different live vaccine against rotavirus that was later withdrawn from the market because of its association with an increased risk of intussusception, a rare, life-threatening type of blockage or twisting of the intestine. Intussusception occurs spontaneously in approximately 1 in 2,000 healthy young infants and children per year, but occurred at an increased rate during the first week or two following vaccination with the previous rotavirus vaccine.
"Although this large study did not show an increased risk of intussusception associated with RotaTeq, given the experience with the previous vaccine, safety of this vaccine will be closely monitored in additional studies conducted after licensure," said Dr. Goodman. The manufacturer has committed to conducting a post-licensure study of approximately 44,000 children. CDC will also conduct a large study designed to rapidly detect any association of intussusception with RotaTeq through its Vaccine Safety Datalink Program, which evaluates vaccine safety in approximately 80,000 U.S. infants every year. In addition, for the first 3 years of licensure, the manufacturer will report cases of intussusception and all serious and unexpected adverse events to FDA within 15 days of receiving them, and all other side effects on a monthly basis.
FDA Approves Vivaglobin
On January 9, 2006, FDA announced the approval of the first immune globulin product for subcutaneous injection for the prevention of serious infections in patients with primary immune deficiency diseases (PIDDs). Vivaglobin, manufactured by ZLB Behring from human plasma collected at U.S.-licensed plasma centers, provides new delivery options for PIDD patients. It is given under the skin (subcutaneously) on a weekly basis using an infusion pump, allowing patients to self-administer the product at home. Some patients develop problems that make chronic intravenous administration of needed medicines difficult, and Vivaglobin may be helpful in providing them with an alternative route of administration.
PIDDs are genetic disorders that affect an estimated 50,000 people in the U.S. and include X-linked agammaglobulinemia (Bruton's disease), common variable immune deficiency, and severe combined immune deficiency (boy-in-the-bubble disease), among others. Patients with PIDD require regular treatment with immune globulin to fight off or prevent potentially serious or life-threatening infections. Other immune globulin products are administered either into the vein (intravenously) or into the muscle (intramuscularly).
"This is an important approval for patients who need lifesaving immune globulin products," said Jesse Goodman, M.D., M.P.H. "This new product provides a unique new treatment delivery option to patients and their physicians."
FDA Approves New Manufacturer of Cytogam
On August 16, 2006, CBER announced the licensure of a new facility and manufacturer, MedImmune, for production of Cytomegalovirus Immune Globulin Intravenous (Human) (Cytogam). Cytogam is a purified antibody preparation made from the plasma of donors with high antibody levels against cytomegalovirus (CMV). Cytogam is used to prevent severe infections with CMV disease in people with kidney, liver, pancreas, lung, and heart transplants. There is no other anti-CMV product licensed in the U.S. and there is no other similar treatment for transplant patients.
The previous sole manufacturer, Massachusetts Public Health Biologic Laboratories, planned to cease production in August. CBER worked with MedImmune to enable timely licensure of Cytogam manufacture at a new facility to avert a shortage of Cytogam for patients in need. Licensure occurred in a timely fashion, precluding any shortage of this important product.
FDA Approves ADVIA Centaur HIV 1/O/2 Assay
On May 18, 2006, FDA approved the first fully automated, random access system for detection of antibodies to HIV-1 group M/HIV-O and HIV-2 in serum and plasma, based on chemiluminescent reactions. The ADVIA Centaur HIV 1/O/2 Assay, manufactured by Bayer Diagnostics, is expected to increase the capability of high-volume testing for HIV in clinical laboratories and to allow for HIV antibodies to be tested along with other analytes in a random access mode. The test is highly sensitive and specific for the detection of antibodies to not only HIV-1, the most common form of HIV in the U.S., but also to the less common HIV-2 and HIV-1 Group O. As with other test systems, this fully automated test system reduces the risk of operator error. There are numerous features to protect the integrity and security of the test system.
Improve Product Quality, Safety, And Availability Through Better Manufacturing And Product Oversight
CBER has emphasized the development and implementation of product safety teams to address areas, such as adverse events, product manufacturing and quality issues, and response to emerging threats. The Center has addressed issues related to safety of human tissue transplants, increased demand for immune globulin products and platelets, as well as risks to the blood supply from Transmissible Spongiform Enchephalopathies (TSE) agents.
Ensuring Safety of Recipients of Donor Tissue
On October 25, 2005, FDA notified the public of its investigation of human tissue recovered by Biomedical Tissue Services Ltd. (BTS) and sent to tissue processors. The tissue was recovered by BTS from human donors, who may not have met FDA donor eligibility requirements, and who may not have been properly screened. The tissue processors voluntarily recalled all unused tissue remaining in inventory, and worked cooperatively with FDA to ensure the implanting physicians whose patients may have received the products were properly notified.
On January 31, 2006, FDA issued an Order to Cease Manufacturing and to Retain Human Cells, Tissues, and Cellular- and Tissue-Based Products (HCT/P) to BTS. This was the first Order that FDA has issued under the HCT/P regulations, which went into effect May 25, 2005. The Order was issued after FDA's inspection of BTS uncovered serious violations of the regulations governing donor screening and record-keeping practices.
On March 2, 2006, FDA issued an update strongly recommending that health care providers inform their patients who received tissue implants prepared from BTS donors, that they may be at increased risk of communicable disease transmission and to offer them testing. This update was based on additional information that called into question the reliability of some of the donor samples submitted for communicable disease testing from BTS tissue donors. In some instances, blood samples did not come from the same donor as the linked tissue. Therefore, the results of communicable disease tests obtained from the blood samples may not correctly reflect the status of the donor. FDA's investigation into BTS activities is ongoing. FDA continues to work cooperatively with tissue processors and appropriate federal, state, and local authorities in this matter.
Donor Referral Services
On August 18, 2006, FDA issued an Order to Cease Manufacturing and to Retain HCT/Ps to Donor Referral Services (DRS). The Order was issued after an FDA inspection, and a concurrent investigation revealed the human tissues recovered by DRS may not have met FDA requirements for donor eligibility and was sent to tissue processors. The tissue processors voluntarily recalled all unused tissue remaining in inventory and worked cooperatively with FDA to ensure the implanting physicians whose patients may have received the products were properly notified. FDA continues to investigate DRS activities, working with the appropriate federal and state authorities in this matter.
On August 30, 2006, FDA issued a public health notification informing the health care community that human tissues recovered by DRS may not have met FDA requirements for donor eligibility and there is a potentially increased risk for infectious disease transmission. FDA and CDC strongly recommended that health care providers inform their patients who received tissue initially recovered by DRS that they may have received tissue from donors for whom adequate donor eligibility determinations were not performed and offer patients access to appropriate infectious disease testing.
Immune Globulin Availability and Demand
During FY 2006, FDA and the Centers for Medicaid and Medicare Services (CMS) received reports that health care providers were having difficulty obtaining immune globulin intravenous (IGIV) for some patients. FDA worked cooperatively with DHHS and the Plasma Protein Therapeutics Association (PPTA) to monitor the IGIV supply and facilitate its availability. Although there does not appear to be a severe product shortage, there have been reports of difficulties obtaining the same product in the same treatment center that patients customarily use. FDA, with the DHHS Office of Public Health and Science (OPHS), contacted responsible officials of all IGIV manufacturers to discuss the market situation. As a result, the average IGIV distribution increased by 12 percent in the last 4 months of 2005, compared with the previous 12-month average. Additionally, PPTA listed hotline numbers on its manufacturers' website for physicians to acquire IGIV for emergency use.
At its July 2006, meeting, the Blood Products Advisory Committee (BPAC) discussed the availability of varicella-zoster immune globulin (VZIG) because of the decision of the Massachusetts Public Health Biological Laboratories (MPHBL) to discontinue manufacturing the products. MPHBL was the only licensed U.S. manufacturer of VZIG, which is used to prevent life-threatening varicella-zoster infections in immune-compromised children and adults exposed to chickenpox. FDA sought the Committee's advice on options for efficacy determination for new BLA applications for VZIG because of concerns about a potential upcoming shortage of this product. FDA is working with sponsors and manufacturers, and has allowed an expanded access IND protocol for VZIG from a new manufacturer, to ensure availability under an IND until a new product is licensed. FDA and OPHS will continue to monitor IGIV availability and work with manufacturers, distributors, and others in the product distribution chain toward increasing availability.
TSE Safety for Biological Products*
Blood Donor Deferral for Transfusion in France
Since 2002, FDA has recommended the deferral of blood donors who resided in the United Kingdom (U.K.) for more than 3 months and elsewhere in Europe for more than 6 months because of a potential dietary exposure to vCJD, as a precautionary measure to prevent its possible transmission through blood. Although 90 percent of vCJD cases have occurred in the U.K., FDA has periodically reviewed and updated its recommendations on donor deferral. At the TSE Advisory Committee (TSEAC) meeting in February 2005, FDA shared with the committee a concern about the growing number of vCJD cases recognized in France. FDA requested advice on whether to recommend deferral of blood donors with a history of blood transfusion in France. A majority of TSEAC members recommended that FDA defer donors transfused in France.
Accumulating evidence suggests the time from exposure to vCJD to the onset of symptoms of vCJD develop may be very long, sometimes exceeding 12 years. The risk of dietary exposure to the bovine spongiform encephalopathy (BSE) agent in France, as in the U.K. and other European countries, has almost certainly decreased in recent years due to efforts to control the BSE epidemic in cattle and to protect food from contamination with the BSE agent. However, an unknown but possibly significant number of potential U.S. blood donors might have already been infected due to residence in France during the peak years of the BSE outbreak in Europe. There have been three reports in the U.K. of probable transmission of vCJD through transfusion of blood cells, including a case of blood collected 3 years before the blood donor showed any sign of illness.
These considerations led FDA to conclude it would be a prudent preventive measure to indefinitely defer whole blood and source plasma donors who have received a transfusion of blood or blood components in France since 1980. For source plasma donors, however, FDA concluded an exception can be made if blood components are collected solely for manufacturing into noninjectable products (e.g., materials used in the in vitro diagnostic test kits). To address this deferral, the draft guidance entitled, "Amendment (Donor Deferral for Transfusion in France Since 1980) to Guidance for Industry: Revised Preventive Measures to Reduce the Possible Risk of Transmission of Creutzfeldt-Jakob Disease (CJD) and Variant Creutzfeldt-Jakob Disease (vCJD) by Blood and Blood Products," was published on August 8, 2006. FDA will continue to monitor the BSE epidemic and re-evaluate the necessity of deferring donors transfused in other European countries. CBER continues to evaluate technologies for clearance of infectious agents, including TSEs, from biological products.
TSE Research and Risk Estimation
FDA has continued to work with manufacturers to determine the role of plasma fractionation in reducing TSE risk. Laboratory studies using model TSE agents have demonstrated TSE infectivity may be reduced by certain plasma fractionation manufacturing steps. Although experimental studies are reassuring, not all products have been thoroughly studied. In addition, it remains uncertain if the models accurately reflect the form of infectivity in blood, which has not been characterized. FDA evaluates submissions containing experimental data on TSE clearance for specific products. Based on such data, FDA has approved labeling for several products. In September 2006, CBER asked TSEAC for advice on whether standardized methods and assessment criteria are feasible and appropriate for determining removal of TSE in the manufacturing processes for certain products made from human plasma. At the same meeting, CBER asked TSEAC for advice on potential approaches and issues to consider when validating candidate screening tests for vCJD and other TSE infections in donors of blood, plasma and human cells, tissues, and cellular- and tissue-based products.
FDA attempts to improve the definition of possible risk to the blood supply posed by vCJD and design approaches to reduce that risk. FDA also conducts collaborative research projects to investigate the role of various abnormal forms of the prion protein in TSEs, to explore the potential for accidental contamination with TSE agents of various cell lines used to manufacture biologic products, and to identify methods for reducing the risk of contamination and cross-contamination with TSE agents.
At the TSEAC meeting in February and October 2005, FDA asked the Committee for advice on a computer-assisted probabilistic model for estimating the potential risk of vCJD to recipients of U.S. licensed human plasma-derived coagulation factor VIII products. CBER has taken the advice of TSEAC to refine the model further, and the results of the study will be presented at a future TSEAC meeting and made publicly available.
Workshop on Testing for Malarial Infections in Blood Donors
On July 12, 2006, CBER organized a workshop to discuss the scientific developments that might support donor testing for malarial infections as a part of predonation screening, or, alternatively, follow-up testing of deferred at-malaria-risk donors to permit a reduced deferral period. Currently, in the U.S., no laboratory test is approved to screen donors for malarial infections and at-malaria-risk donors are identified by donor questionnaires. Although donor deferral polices have been effective in reducing the incidence of transfusion-transmitted malaria (TTM) (16 cases of TTM in the last 15 years), this approach has led to the loss of more than 120,000 otherwise suitable blood donors each year. The outcome of the discussion at the workshop revealed the problems associated with malaria test sampling and sensitivity issues, and concluded nucleic acid-based tests are not ready for use in screening of blood donors for malaria parasite infections. However, several European countries, Australia, and New Zealand now test deferred at-malaria-risk donors, which shortens the deferral period for some donors without malarial antibodies. The antibody based test, used in those countries, detects antibodies to only 2 of 4 human Plasmodium species (Plasmodium falciparum and Plasmodium vivax), allowing the earlier re-entry of 97% of the otherwise deferred donors. Workshop participants discussed the merits of such antibody-based tests and possible algorithms of implementation in the U.S. blood-banking industry. FDA is currently discussing malaria antibody test development and donor testing algorithms with various stakeholders, including the blood-banking industry.
Transform Administrative Systems And Infrastructure To Support FDA Operations
CBER has focused on the use of sound management practices and information technology to continue supporting and enhancing the mission of the Center. These efforts include international outreach and collaboration, improving review processes and harmonisation, and management initiatives.
Globalization of Public Health and Product Development: International Activities Highlights
World Health Organization (WHO) and Pan American Health Organization (PAHO) Activities
CBER continues in its second 4-year term as a designated PAHO/WHO Collaborating Center for Biological Standardization, a term that ends October 2007. The Center is looking ahead to what shape its third term might take as it begins discussions with PAHO and WHO on a set of mutually agreed upon Terms of Reference. Through scientific expert consultations and laboratory collaborations, CBER staff contribute their expertise to the WHO's mission to "…develop, establish and promote international standards for biological products," as well as provide leadership in addressing the many and varied new challenges that routinely seem to arise in the world of biological products.
In FY 2006, CBER staff participated in more than 35 consultations spanning such topics as the following:
- immunological correlates of protection induced by dengue vaccines
- requirements for diphtheria, tetanus, acellular pertussis vaccines
- variola virus research; requirements for cell substrates in the production of biologicals
- stability assessment of vaccines; global monitoring of adverse events following immunization; critical regulatory issues associated with human tissue transplantation
- international nonproprietary names for gene therapy products
- quality, safety, and efficacy recommendations for serogroup A meningococcal conjugate vaccines
- specifications and validation of HIV/AIDS diagnostic technologies
- HPV guidelines; and tuberculosis vaccine challenges
CBER continued to provide leadership in key strategic committees and forums of WHO, including the Global Vaccine Safety Advisory Committee, the Expert Committee on Biological Standardization (ECBS), the Immunization Strategic Advisory Group of Experts, and the Global Vaccine Research Forum. CBER also actively participated in WHO's 12th International Conference of Drug Regulatory Authorities (ICDRA), a global biennial conference hosted by individual country health authorities. The April 2006, ICDRA was hosted by the Government of Korea. Of note for the 2006 ICDRA was the convening of an open, 2-day plenary session in advance of the conference with the theme, "Improving World Health through Regulation of Biological Medicines."
In FY 2006, CBER initiated active engagement with the recently formed WHO Developing Country Vaccine Regulator Network (DCVRN). CBER provided staff and support for a vaccine workshop/symposium held in conjunction with DCVRN in November 2005, in Bangkok, Thailand, addressing HIV vaccine development and associated regulatory issues. CBER sent staff to a DCVRN Regulatory Forum entitled, "Clinical Evaluation of New Typhoid Vaccines," held in Seoul, Korea. In addition, CBER was active in launching a new WHO initiative with the WHO Africa Regional Office (AFRO), which is expected to lead to biannual meetings of an AFRO Vaccine Regulatory Forum (AVAREF). During the inaugural meeting in Accra, Ghana, in September 2006, CBER staff provided their expertise in regulatory considerations with respect to clinical trials expected to take place in Africa specific to malaria, meningitis, HIV, and rotavirus candidate vaccines. The vision for AVAREF is to serve as a source of expertise for countries that have to make regulatory decisions for which they are not prepared. Also, another part of the vision for AVAREF is to become a forum in which countries can discuss issues with peers as a means of building on the expertise available in the region, and strengthening the capacity of countries with less mature regulatory frameworks.
In 2004 and 2005, the ECBS first recommended, then later supported, the further development of a framework concept for a Blood Regulatory Network. The vision was to establish a group of experts in the regulation of blood to develop detailed discussions of issues (e.g., vCJD), even when global approaches were not fully harmonised. CBER was active in the development of this framework, which is expected to be presented to the ECBS in early FY 2007.
Global Collaboration for Blood Safety
CBER continued to support the partnership of the Global Collaboration for Blood Safety (GCBS), with CBER's Director of OBRR serving his fourth year as chair. The GCBS was created by WHO in 2000 to implement World Health Assembly Resolution 48.27 (1995), which contained a commitment to international collaboration for blood transfusion safety. According to its terms of reference, GCBS was constituted as "a voluntary partnership of internationally recognized organizations, institutions, associations, agencies and experts from developing and developed countries sharing the expertise, identifying problems, seeking solutions and working toward the common goal of global blood safety as equal collaborative partners. WHO is a participant of GCBS and also provides its secretariat." The GCBS is a forum that facilitates international collaboration in blood safety and availability through dialogue, nonbinding recommendations, and cooperative work. The sixth general meeting of the GCBS was held in Bangkok, Thailand, in November 2005. A spectrum of organizations, institutions, and associations with international interests related to global blood safety attended.
International Conference on Harmonisation (ICH)
ICH is a unique project that brings together the regulatory authorities and pharmaceutical industry experts from Europe, Japan, and the U.S. to discuss scientific and technical aspects of product registration. CBER joins CDER as members of ICH Steering Committee and provides technical representation on the ICH Expert Working Groups, Implementation Working Groups, Informal Discussion Groups, and Brainstorming Groups.
Of particular note, specific to CBER, was the work of its experts in the Gene Therapy Discussion Group (GTDG). A public, 1-day workshop on "Oncolytic Viruses" was held in conjunction with the ICH Steering Committee meeting in Chicago, IL, in November 2005. Objectives of the workshop were to identify and discuss issues relevant to clinical development of oncolytic viruses, including safety. Over the course of the following ICH meetings, the GTDG discussed the outcomes of the workshop and began to draft an ICH Considerations Document on the topic using the workshop as its basis. Fiscal year 2006 saw the initiation of several new topics for harmonisation, including "Pharmaceutical Quality Systems" (Q10), "Pharmacogenomics" (E15), and "Development Safety Update Reports." There was also the revision of several previously harmonised preclinical (safety) guidelines: "Nonclinical Safety Studies for the Conduct of Human Clinical Trials" (M3), "Genotoxicity" (S2A/B), and "Dose Selection for Carcinogenicity Studies of Pharmaceuticals" (S1C). Finalized guidelines included, "Pharmaceutical Development" (Q8) which describes what should be submitted to a regulatory authority in the relevant section of the Common Technical Document [ICH topic M4], and "Quality Risk Management" (Q9) which provides principles and examples of quality risk management that can be applied to all aspects of developing a medicinal product, submitting to a regulatory authority, and manufacturing site inspections.
New ground was broken as the ICH Steering Committee explored options for the development of technical messaging standards developed exclusively within ICH. Discussions were held on the potential future use of standard development organizations (SDOs) for the development of technical standards relating to the electronic transmission of regulatory information.
International Partnering
Fiscal Year 2006 saw a maturation of FDA's use of bilateral information sharing agreements with strategic foreign regulatory counterparts to effect and enhance regulatory cooperation. Several new agreements were finalized, and earlier agreements saw increased operational exchanges. CBER both initiated and responded to requests for information exchange from counterpart agencies in contexts ranging from inspectional issues, to data interpretation, to surveillance signals. Other activities undertaken included the hosting of two separate longer-term visits from SwissMedic, the regulatory body in Switzerland. The first visit focused on our regulation of blood and blood products, and the second visit focused on our regulation of gene therapies. Pursuant to an information sharing agreement, CBER staff continued their participation in on-going Agency dialogues with the European Medicines Agency (EMEA) in the areas of pharmacogenomics, pediatrics, pandemic influenza vaccines, and cancer therapeutics, as well as participated on-site in several expert meetings held at the EMEA. CBER partnered with Health Canada and WHO in FY 2006 to hold two meetings of global regulators to tackle regulatory challenges with respect to pandemic influenza vaccines. Utilization of these agreements has proven to be value-added to the range of the Center's responsibilities.
Increasingly and more routinely, CBER sought the participation of international experts, both regulatory and nonregulatory, in the Advisory Committee meetings and specific technical workshops (e.g., the workshop on Molecular Methods in Immunohematology held in September 2006.) CBER continues its active involvement in a number of international scientific working forums, such as the International Working Group on the Standardization of Genomic Amplification Techniques (SoGAT) for the Virological Safety Testing of Blood and Blood Products. In February 2006, CBER co-sponsored with the Japanese government the Ninth US-Japan Cellular and Gene Therapy Conference on Genomics and Proteomics Technology in Biomarker Discovery.
Pharmaceutical Inspection Cooperation Scheme (PIC/S)
One element of FDA's "GMPs for the 21st Century" initiative in 2003-2004 was to pursue membership in the Pharmaceutical Inspection Cooperation Scheme (PIC/S). PIC/S is an international association of national regulatory authorities with inspectional and oversight responsibilities for medicinal products. The primary goals of PIC/S are to strengthen cooperation between the participating authorities, provide a framework for the exchange of necessary information and experience with respect to inspectional matters, coordinate mutual training of inspectors, and generally pursue harmonisation of technical standards in the inspection of the manufacture of medicinal products. The vision for FDA's participation in this cooperative arrangement is one in which inspectional information received from other regulators can serve as a component of a larger risk-based strategy to our inspection program. In FY 2005, FDA submitted its application to join the PIC/S, an application of substantial detail to which CBER made significant contributions. FY 2006 has seen the continued interaction between FDA and PIC/S to provide clarification of our inspectional system and regulatory framework as the membership process moves forward. CBER continues to contribute significantly to this effort.
Global Harmonisation Task Force
The Global Harmonisation Task Force (GHTF) was conceived in 1992 in an effort to respond to the growing need for international harmonisation in the regulation of medical devices. Primarily an engagement with FDA's CDRH and ORA, GHTF is also relevant to CBER due to CBER's regulatory oversight of certain devices (e.g., blood donor screening tests). CBER staff work closely with the CDRH representatives to the five GHTF expert study groups and contribute as needed.
International Outreach
CBER staff participate in international forums to provide outreach to both the regulatory and scientific global communities. A sampling of these interactions shows the breadth of these efforts; in FY 2006 CBER staff performed the following:
- discussed standardization of microparticle assays at the Biomedical Excellence for Safer Transfusion Collaboratives 31st Semiannual Meeting held in Spain
- participated in a session on vaccine Good Manufacturing Practices (GMPs) at a conference held in Germany on Current Challenges of GMPs in Europe
- participated in the 12th Annual Meeting of the International Society for Cellular Therapy held in Germany
- participated in the 13th International Meeting on Hepatitis C and Related Viruses held in Australia
- participated in the 16th International Congress of the International Organization for Mycoplasmology held in the United Kingdom
- presented at the 6th International Conference on Typhoid Fever and Other Salmonelloses held in China
- participated in the European Centre for the Validation of Animal Models (ECVAM) Workshop on the Consistency of Production Approach and its Potential to Reduce Animal Tests in the Quality Control of Vaccines held in Italy
- participated in the 13th PIC/S Expert Circle on Human Blood and Tissue held in the Netherlands
CBER scientists have also successfully established a number of research collaborations that include international partners, such as the following: Russian and ex-Soviet scientists under the U.S. government's Biotechnology Engagement Program to explore critical public health challenges, the Institute of Neurological Research in Argentina to evaluate potential TSE diagnostic tests, the Institute of Animal Health in Edinburgh, Scotland similarly on a TSE project, and WHO, Program for Appropriate Technology in Health (PATH), and India on a Meningitis Vaccine Project.
Review Management Initiatives
The Review Management staff is active in improving CBER's review processes for biologics, drugs and devices, as well as the associated information technology (IT) support. The Review Management staff works closely with FDA on initiatives to harmonize business processes across FDA and other centers, especially CDER, CDRH, and DHHS. This harmonisation includes database harmonisation wherever possible/feasible.
Agency Harmonisation Initiatives
These initiatives include participation in the Agency's Bioinformatics Board. This Board was approved by FDA's Senior Management in February 2006. The Board will control FDA's business process development relative to planning and management of electronic data, systems, and tools to support regulatory decisions, and communications across all FDA operations. CBER staff serve in various capacities on the Board and its subcommittees (Business Review Boards), including Co-chair of the Pre-Market Business Review Board and the Data Standards Development Lead for the Post-Market Review Board.
FDA harmonisation initiatives also include development of an Enterprise Architecture (EA) in which CBER fully participates. The initiative includes mapping CBER business processes to the EA model and evaluating them for commonality to facilitate future business system automation. Review Management has engaged the new Agency EA contractor to continue this effort.
Quality Systems Initiatives
CBER is also working closely with the Agency on the development of quality systems within the Agency and the Center. These systems are under development in coordination with CDER as part of the PDUFA III reauthorization. Examples of these systems are the development of a quality system for Chemistry, Manufacturing and Controls (CMC) review, as well as templates for action letters, meeting minutes, and regulatory reviews. Both of these initiatives are led in the Center by Review Management staff.
For FDA-PDUFA III Quality Systems initiatives, CBER is the lead on facilitating the development of an interactive, Web-based training for reviewers. Training is intended to improve regulatory review using CBER's regulatory database systems. CBER will provide and coordinate the training content and participate in developing the Postmarketing Commitment (PMC) Project. CBER participates in this CBER-CDER working group to monitor progress of the contract used to evaluate the consistency of processes for requesting and reviewing reportable PMCs (clinical safety, effectiveness, pharmacology, and nonclinical toxicology) and the outcomes reports as required by section 506B of FDAMA.
In addition, CBER collaborates with the Office of Planning in the Office of the Commissioner to implement initiatives for PDUFA. These projects include analysis of First Cycle Review Approvals, IT systems management, and harmonisation across the Centers.
Electronic Standards Initiatives
CBER is active in Agency electronic standards development. These activities include participation in FDA Data Standards Council and its subgroups: the Adverse Event Workgroup (Chair) and the Regulated Product Submission (RPS) Group (CBER Lead and Project Coordinator). All Centers receive applications from industry containing information on regulated products. The purpose of the RPS standard is to develop a comprehensive set of headings and hierarchy for the organization of these applications. The standard is based on the Common Technical Document developed by CDER and CBER and adopted by the ICH.
Another specific example of CBER involvement in electronic standards development is the Health Level Seven (HL7) Patient Safety Special Interest Group (Cochair) initiative for DHHS. This initiative provides for the creation of HL7 patient safety and public health reporting in a standardized electronic format (messaging) for communication, including Individual Case Safety Report (ICSR) and Generic Patient Safety Incident Notification. The ICSR communication format will improve the quality of direct reports from health care providers by using a standardized electronic reporting format for all FDA-regulated products. The ICSR will serve as a data collection template to support redesign of the MedWatch website. A draft standard for trial use was approved by HL7 in May 2006. FDA expects to conduct limited pilot testing with various industry partners in FY 2007.
CBER is also coordinating the Clinical Data Interchange Standards Consortium (CDISC), Study Data Tabulation Model (SDTM) pilot. The project goal is to establish a standard electronic format for industry to use when sending study data to FDA. CBER is working with Merck to evaluate the CDISC-SDTM format for vaccine submissions. The initial pilot of the CDISC-SDTM incorporated use of review tools (developed by CDER) to view submissions, as well as to identify areas that will require further CBER standards development for CDISC and HL7.
Outreach Initiatives
CBER continues to participate with FDA and DHHS on outreach efforts and harmonisation efforts. In addition to conferences and presentations, this area includes work with the Federal Health Architecture (FHA) Public Health Surveillance (CBER Liaison and Patient Safety Subgroup Chair), American National Standards Institute Health Information Technology Standards Panel (HITSP) (CBER Liaison), the Pharmaceutical Research and Manufacturers of America (PhRMA) Electronic Regulatory Submissions Working Group, the U.S. Pharmacopeia (USP), and the ICH. CBER staff serve on the ICH Steering Committee, the ICH Quality Initiatives, and the E2B, M5 Expert Work Groups, and the Controlled Terminology Maintenance Pilot Work group.
CBER Review Business Process Efforts
CBER continues to develop ways to improve the efficiency of the review business process. CBER's Review Management Coordinating Committee (RMCC) works to achieve consistency and harmonisation of the review process across the Center. The RMCC consists of senior management and leaders of each Review Office in the Center. The RMCC makes certain that all of the Center's regulatory Standard Operating Procedures and Policies (SOPPs) are consistent with CBER's Managed Review Process.
RMCC also recommends changes to the review business process to maximize use of the electronic systems that support the regulatory effort. As part of this effort, RMCC subcommittees include a consolidated business process working group and a layered approach for implementation of IT regulatory systems changes, including a consolidated change control board to address impact across the various CBER IT systems. It is expected the closer interaction between the business community and the IT community will lead to improved systems with a greater level of integration. Consolidation of the business process working groups will allow members to more effectively identify issues and solutions that cross boundaries of work areas. Similarly, consolidation of the individual regulatory systems working groups and change control boards will eliminate redundancy and enable decisions to be made that will allow all systems to move forward in the most efficient manner. One example of an initiative put forward is the establishment of procedures for the use of electronic signatures for all CBER-generated regulatory documents. This initiative will utilize the new framework in coordination with changes to the business processes and the regulatory databases.
CBER works to develop enhancements to current regulatory systems and to develop new systems. Current initiatives include presubmission tracking for meeting packages prior to the submission of either an investigational or marketable application. With the new regulatory system in place, product offices will enter presubmission information and presubmission links to the associated meetings more efficiently. This Web-based system will track presubmissions for all application types received in CBER.
CBER focuses on consistent review processes throughout the Center for manufacturing processes. The CMC-CC (CMC Coordinating Committee) and its subcommittees work closely with FDA in the development of guidance documents, rulemaking, and Center SOPPs to achieve harmonisation and consistency with other Centers on projects, such as Rapid Microbial Methods (RMM). CBER is also involved in FDA activities, such as the Process Analytical Technologies (PAT), the Pharmaceutical Quality Council, and the Current Good Manufacturing Practices (FDA Co-chair) committees. In addition, CBER works with the review community on the development of guidance documents. This includes topics, such as process validation, cooperative manufacturing, electronic submissions; current, good manufacturing practice for INDs, and chemistry, manufacturing, and controls.
CBER is working to further the transition from review of paper to electronic submissions. To achieve this goal, consistency among the regulatory databases and the data entered was enhanced. Highlights of the electronic review process include:
- Coordinate the conversion from paper to electronic forms for data entry and routing/review of IND/ Investigational Device Exemption (IDE)/Master Files (MF)/Emergency Use Authorization (EUA) submissions. To achieve this goal, CBER provided the requirements for Biologics IND Management Systems (BIMS)/Biologics Investigational and Related Applications Management System (BIRAMS) changes to accommodate the conversion and provide training to office staff on the use of electronic forms.
- Work closely to coordinate ongoing streamlining of review procedures, including handling of consult review and improved integration with the electronic document room (EDR).
- Data standardization and enhancement of the BIRAMS: these enhancements simplify data entry and updates, as well as improve the quality of the data. This initiative is part of the overall data improvement plan for CBER's regulatory systems to help the review community manage their reviews and submissions more efficiently.
- Work to finalize guidance and establish administrative procedures on the submission of lot release protocols in electronic format.
CBER continues to keep the review community updated with monthly Review Management Updates sessions. Each session begins with a review of the rules, guidances, and SOPPs recently published and what their impact will be on the review community.
Quality Assurance
CBER focuses efforts on dispute resolution and providing an objective resource for the ongoing review and evaluation of Center programs and operations.
As part of CBER's Strategic Plan for 2007, the Center has reaffirmed its commitment to ensure quality in the performance of all core functions. To this end, a variety of quality assurance activities are being conducted for many of CBER's research, review and compliance activities.
CBER supports quality assurance activities, assists in the identification of problem areas, and helps to identify potential solutions. CBER also monitors implementation and the impact of changes in the Managed Review Process and provides an ongoing evaluation of CBER's efforts to meet relevant PDUFA performance goals for specific product categories. In FY 2006, as part of CBER's ongoing quality assurance efforts, three Clinical Hold Oversight Committee meetings were held, and one more meeting is planned before the end of calendar year 2006. In addition, CBER has initiated a pilot program to automate internal auditing of administrative activities associated with completed action files.
The Lab Quality System Team continued to lead the Center's efforts to gain laboratory accreditation. Their FY 2006 accomplishments include the following: implementing an electronic sample tracking system, piloting an internal auditing program for laboratories, updating the Center Laboratory Quality Policy Manual, as appropriate, during this design and implementation phase, and continuing efforts toward completion of the Manual.
It is anticipated that this laboratory accreditation effort will continue throughout FY 2007. In addition, CBER is participating in FDA Quality Resource and Guidance Team, a subcommittee of the FDA Management Council that is providing guidance on the internal application of quality systems; and FDA-PDUFA Quality System Group, overseeing the use of PDUFA funds toward the internal application of quality systems within FDA.
CBER's Ombudsman was established to investigate and act on complaints regarding the CBER regulatory review processes and to provide an effective informal process for resolution of regulatory or scientific problems that cannot be resolved by other means. In instances where an informal process is not adequate, the CBER Ombudsman may serve as the mediator or arrange for mediation to conduct the formal dispute resolution process as defined by FDAMA.
In FY 2006, CBER received approximately two informal requests per week for assistance, primarily from outside FDA. No formal dispute resolution requests were received. Of the informal requests, twenty-four required a substantial level of intervention or mediation and three were internal to FDA.
In addition, the CBER Ombudsman handles complaints and questions about inter- and intra- center product jurisdiction and serves as a member of the Tissue Reference Group. With respect to inter-center jurisdiction in FY 2006, more than 60 Requests for Designations were received, 16 of these requests were specifically assigned to CBER for evaluation. Many of these requests related to combination products that included a biologic and device, biologic and drug, or drug and device component, as well as products that contain a human tissue component in combination with a regulated article. Informal jurisdiction questions received from inside and outside FDA averaged fi ve to ten per month.
Finally, CBER provides oversight of the review of 513(g) Requests for Information. A CBER response to this type of request will state if the product in question meets the definition of a device as defined in the Federal Food, Drug, and Cosmetic Act, and, if so, whether the device is subject to regulation under the Act. This was implemented in response to the increasing number and complexity of 513(g) requests to CBER. There were six 513(g) requests to CBER in FY 2006: two were deemed not to be devices defined by the Act; one was deemed a device, but not subject to regulation; one was deemed to be regulated as a class II device; and two are currently under review.
Management Systems Initiatives
In FY 2006, CBER introduced a variety of initiatives to modernize the operations and business practices of the Center. In particular, CBER was successful at converting to the BIRAMS, the first major upgrade of the system in 14 years. BIMS/BIRAMS is the mission-critical database system that tracks submissions, communications, and decisions related to investigational studies on human subjects. Regulatory applications tracked in this system include INDs, IDEs, master fi les, and EUAs. The information tracked in this system represents nearly 50 percent of CBER's regulatory review workload. Data are time-critical and sensitive. The system interfaces with twelve other systems.
As part of an effort to standardize BIRAMS data and to prepare for the implementation of BIRAMS and its new sponsor/contact information model, CBER reviewed, edited, and verified nearly 61,000 records and consolidated them down to 22,000 records before the data migration. As a result, the current sponsor and contact information in BIRAMS are more accurate and consistent.
IT and System Enhancements
CBER provides value-added technology solutions in support of our regulatory processes. CBER is committed to using the best software practices and optimizing its resources to increase and maintain system functionality based on program priorities. The strong relationship between the business process group and CBER continues to grow. To ensure that development and changes made to business processes can be supported and enhanced by IT, CBER supported the reorganization of the structure of our system change control boards and participates in the business process working group. We look forward to continued process improvements and working together to the better use of IT to facilitate the regulatory processes.
CBER strives to implement industry best practices and use technology effectively to support our business community by providing automated and integrated tools, databases, and systems that enable staff to fulfill CBER's mission. One industry best practice is the Capability Maturity Model (CMM) for project management of software development projects. CBER received certification for CMM level 2 rating for its Project Management Group in December 2005. CBER is now adopting the FDA's Office of the Chief Information Officer (OCIO) requirement that Centers and Offices use the "FDA Investment Life Cycle" procedures that involve the use of standardized templates and stage gates.
Working closely with the Office of Information Technology Shared Services (OITSS) Employee Resource & Information Center (ERIC) to provide assistance to CBER customers, CBER resolved more than 100 ERIC trouble calls this year. CBER continues its support for CDER employees who utilize CBER systems, processing more than 80 account requests to access CBER regulatory applications systems in FY 2006.
To support the Pandemic Flu Program, CBER coordinated several efforts, including the selection and procurement of equipment necessary for new staff. CBER collaborated with FDA's OCIO and OITSS in the development of plans and solutions to ensure remote access during a crisis, database failure, and disaster recovery. These solutions are scheduled for implementation next year.
eSystems
CBER maintains and enhances several systems that support the electronic submission of information, including the "CBER On-Line" system to facilitate the submission of the following three FDA forms:
- Form FDA 2830 for Blood Establishment Registration and Product Listing
- Form FDA 3356 for Establishment Registration and Listing for Human Cells, Tissues, and Cellular and Tissue-Based Products (HCT/Ps)
- Form FDA 3486 for Biological Product Deviation Reports (BPDRs).
Form Number |
% Electronic in FY 2005 |
% Electronic in FY 2006 |
|---|---|---|
Form FDA 2830 |
70 |
78 |
Form FDA 3356 |
47 |
49 |
Form FDA 3486 |
63 |
74 |
There has been an increase of several hundred additional facilities using the registration models and several thousand additional BPDRs entered online this year, compared with FY 2005. Through this system, the information is sent directly to the appropriate coordinators. Because it is already in an electronic format, this provides efficiency in time and resources for CBER.
Query systems were also created by CBER for use by the general public, FDA's ORA, and agencies searching registrations for Blood Establishment and Human Cells, Tissues, and Cellular- and Tissue-Based Products (HCT/ Ps). These systems reduce the number of calls to the Registration Coordinators thereby allowing their efforts to be focused on processing and analysis of received information.
EDR and FDA Gateway
CBER's EDR functions as an electronic library for reviewers, routing and storing electronic submissions of IND, BLA, NDA, 510(k), PMA, regulatory correspondence, and other CBER data. The EDR is also integrated with the CBER regulatory databases to allow for advanced searches. In FY 2006, more than 2,000 electronic submissions, including BLAs, INDs, and amendments, reports and correspondence, were accepted and installed.
EDR enhancements for FY 2006 included the completion of a major technology enhancement to its architecture, as well as adding full-text searching capabilities to all stored electronic applications, integration with the new BIRAMS. Enhancements also included consolidation of the CBER Secure E-mail System into a single FDA Secure E-mail System, and the capability that allows Acrobat 7 forms to be processed with electronic submissions.
The FDA Gateway, a single portal for submittal of both PMAs and adverse events, was placed into production in May 2006. We are proud that CBER was able to provide the Project Manager and the Lead Technical Representative for this Agency project. CBER received more than 100 premarket electronic submissions via the Gateway this year. CBER has started to collect the requirements necessary to automate the loading of documents received via the Gateway into the CBER EDR.
Other System Enhancements
The BIMS was redesigned and renamed the BIRAMS. The BIRAMS supports high-level tracking and summarization of CBER regulatory efforts associated with INDs, MFs, IDEs and EUAs. Modifications to the BIRAMS system included improvements to the data quality in the system and increased functionality of handling investigational submissions. Other changes included modifications to the electronic forms (electronic Original Submission and electronic Preliminary IND Amendment Slip [ePIAS]), as well as the addition of the electronic Review Process (eReview sheet). The eReview sheet made reviews instantly available in the CBER EDR, improved the managed-review cycle, allowed supervisors the possibility of concurrence and the possibility to flag items for follow-up, reduced paper handling, and saved resources. The eReview sheet allows the reviewer to view BIRAMS-related documents in the EDR without having to leave the BIRAMS system.
This major upgrade of the system was implemented in June 2006, and is the first of three phases. This upgrade encompassed a complete restructuring of the underlying database for sponsor, contact, and IND characteristics data. It also included a mass data cleanup of the sponsor and contact information. This implementation required the coordination with twelve other systems that interface with BIRAMS. All interfacing systems had to be implemented at the same time as the interface changes with the new BIRAMS structure.
Phase II enhancements will further improve the database structure and data quality. The database structure will incorporate the blending of two statuses into one and the redesign of the communications module, which will be usable by other systems. Phase III, the last revision of the system before going into a mandated maintenance stage, will be a technology upgrade. Because of the successful on-time implementation of phase I, these next two phases will be completed in FY 2007.
Benefits to the Center for this conversion are substantial. The underlying database will be normalized and stable, making future enhancements less costly. The system is now fully compatible with all of the interfacing systems, as well as the several electronic forms used in the document management and review processes. Data quality, which is critical to the system's usefulness to the reviewers, has improved significantly because of standardization and cleanup of the sponsor/contact information and other data subsets.
The Regulatory Management System for the BLA (RMSBLA) provides an automated system to support the tracking of BLAs, their review, and their associated data. CBER successfully implemented three major software upgrades to the RMS-BLA system comprising more than 100 user- and programmer-generated change requests, 300 data change requests, several performance-related enhancements, and 15 special report requests. Among these enhancements were modifications to facilitate more Center-specific reporting and processing resulting from the transfer of biotech products to CDER, a reviewer "In Box" navigable list of submissions with which the user is associated, more automated letter generation, work needed in preparation for the GS review tool for electronic Common Technical Document (eCTD) electronic submissions, integrated routing request subsystem, and additional search and reporting capabilities. In addition, an Annual Report review template within the RMS-BLA system to aid in the review, tracking of, and reporting on Post-Marketing Commitments (PMCs) was developed.
The Lot-Distribution Database (LDD) system automates the collection of lot-distribution data, as required by regulation. Enhancements in FY 2006 include improvements to handling of packaged products (two or more products sold together) and changes to simplify the data-entry process, requiring extensive contact with the manufacturers who submit LDD data. Planning has begun for future implementation to replace the largely manual LDD data-entry process with a fully automated process using the FDA Gateway.
Security
IT Security Programs focus on management controls to ensure the integrity of systems. All CBER business systems were certified and accredited for operations in FY 2005-2006. CBER EDR was recertified and accredited as a result of a major upgrade to the system this year. Alignment of the security reporting structure with FDA's Enterprise Architecture and Capital Planning and Investment Control systems designations was also achieved. In support of the Federal Information Security Management Act of 2002 (FISMA), self-assessments and Privacy Impact Assessments (PIA) for nine systems were completed this year. In addition, an eAuthentication assessment was provided to meet eGovernment Act of 2002 requirements. On a quarterly basis, mitigation reports known as Plan of Actions and Milestones (POA&M) are developed, and a review process for account management of CBER's business applications was initiated to ensure proper user roles and access.
Contact Us
Consumers and Health Professionals
800-835-4709 or 301-827-1800
Fax: 301-827-3843 / octma@cber.fda.gov
www.fda.gov/cber
Manufacturers and Distributors
800-835-4709 or 301-827-1800
fax 301-827-3079/ matt@cber.fda.gov
Ombudsman
301-827-0379 / fax 301-827-2920
sherry.lard@fda.hhs.gov or howard.balick@fda.hhs.gov
Freedom of Information Act (FOIA) Requests
800-835-4709 or 301-827-1800 www.fda.gov/opacom/backgrounders/foiahand.html
VAERS
800-822-7967 / fax 877-721-0366
info@vaers.org / http://Vaers.hhs.gov/ http://Vaers.hhs.gov
Genetic Modification Clinical Research Information System (GeMCRIS)
www.gemcris.od.nih.gov
MEDWATCH
800-FDA-1088 or 301-827-7240
fax 301-827-7241 or 800-FDA-0178
www.fda.gov/medwatch/index.html
Biological Product Deviation Reporting
301-827-6220 / bp_deviations@cber.fda.gov
www.fda.gov/cber/biodev/biodev.htm
E-Mail Distribution Lists
To subscribe, click on www.fda.gov/cber/pubinfo/elists.htm
- BLOODINFO
Includes all blood-related documents. - CBERINFO
Includes TISSUEINFO, BLOODINFO, and all other new CBER documents. - TISSUEINFO
Includes all tissue-related documents
Appendix A (CBER Publications)
- Gaines AR. Disseminated intravascular coagulation associated with acute hemoglobinemia or hemoglobinuria following Rh(o)(D) immune globulin intravenous administration for immune thrombocytopenic purpura. Blood. 106(5):1532-1537 (2005). Epub May 5, 2005.
- O'Connell KA, Wise RP, Lozier JN, Braun MM. Recombinant factor VIIa and thromboembolic events (reply letter). JAMA. 296(1):44 (2006).
- O'Connell KA, Wood JJ, Lozier JN, Wise RP, Braun MM. Thromboembolic adverse events after use of recombinant human coagulation factor VIIa. JAMA. 295(3):293298 (2006). [PMID:16418464].
- Lee S, Hu J, Tang S, Wood O, Francis K, Machuca A, Rios M, Daniel S, Vockley C, Awazi B, Zekeng L, Hewlett I. Evaluation of FDA licensed HIV assays using plasma from Cameroonian blood donors. J Med Virol. 78(S1):S22-S23 (2006).
- Orton SL, Stramer SL, Dodd RY. Self-reported symptoms associated with West Nile virus infection in RNA-positive blood donors. Transfusion. 46(2):272-277 (2006).
- Khan AS, Kumar D. Simian foamy virus infection by whole-blood transfer in rhesus macaques: potential for transfusion transmission in humans. Transfusion. 46(8):1352-1359 (2006).
- Laassri M, Lottenbach K, Belshe R, Rennels M, Plotkin S, Chumakov K. Analysis of reversions in the 5'-untranslated region of attenuated poliovirus after sequential administration of inactivated and oral poliovirus vaccines. J Infect Dis. 193(10):1344-1349 (2006).
- Cutrone R, Lednicky J, Dunn G, Rizzo P, Bocchetta M, Chumakov K, Minor P, Carbone M. Some oral poliovirus vaccines were contaminated with infectious SV40 after 1961. Cancer Res. 65(22):10273-10279 (2005).
- Izurieta HS, Haber P, Wise RP, Iskander J, Pratt D, Mink C, Chang S, Braun MM, Ball R. Adverse events reported following live, cold-adapted, intranasal infl uenza vaccine. JAMA. 294:2720-2725 (2005).
- Taylor DR. Obstacles and advances in SARS vaccine development. Vaccine. 24: 863-871 (2006).
- Schaecher K, Kumar S, Yadava A, Vahey M, Ockenhouse CF. Genome-wide expression profi ling in malaria infection reveals transcriptional changes associated with lethal and non-lethal outcome. Infect Immun. 73:6091-7100 (2005).
- Darnell ME, Taylor DR. Evaluation of inactivation methods for severe acute respiratory syndrome cornonavirus in noncellular blood products. Transfusion. 46(10):1770-1777 (2006).
- Varricchio F, Reed J, VAERS Working Group. Follow-up study of medication errors reported to the vaccine adverse event reporting system (VAERS). South Med J. 99(5):486-489 (2006).
- Fink DW, Bauer SR. Stem cell-based therapies: FDA regulatory considerations. Essentials of Stem Cell Biology. 503-511 (2005).
- Manickan E, Smith JS, Tian J, Eggerman TL, Lozier JN, Muller J, Byrnes AP. Rapid Kupffer cell death after intravenous injection of adenovirus vectors. Mol Ther. 13(1):108-117 (2006).
- Malarkey M, Solomon R, Witten C, Bloom E, Wells M, Braun M, Wise R, Zinderman C, Jernigan DB, Kuehnert MJ, Srinivasan A, Wang S. Brief Report: Investigation into recalled human tissue for transplantation --- United States, 2005-2006. MMWR. 55:564-566 (2006).
- Wood JJ, Malek MA, Frassica FJ, Polder JA, Mohan AK, Bloom ET, Braun MM, Coté TR. Autologous cultured chondrocytes-related adverse events reported to the US Food and Drug Administration. J Bone Joint Surg. 88:503-507 (2006).
- Arcidiacono-Horvath JA, Porter CM, Bloom ET. Human NK cells can lyse porcine endothelial cells independent of their expression of Gal (1,3)-Gal ( Gal) and killing is enhanced by activation of either effector or target cells. Xenotransplantation. 13:318-327 (2006).
- Gemeniano M, Mpanju O, Salomon DR, Eiden MV, Wilson CA. The infectivity and host range of the ecotropic porcine endogenous retrovirus, PERV-C, is modulated by residues in the C-terminal region of its surface envelope protein. Virology. 346:108-117 (2006).
- Morgan DA, Argaw T, Wilson CA. Umbilical cord blood stem cells induced into myeloid, erythroid, or megakaryocytic pathways are susceptible to infection by porcine endogenous retroviruses (PERV). Blood. 106(11):4189 Part 2 (2005).
- Ng T-H, Hshieh P. Can we recruit additional subjects for a failed study and still maintain the type one error rate? Proceedings of the Annual Meeting of American Statistical Association. (2006).
- Finkleman MA, Lempitski SJ, Slater JE. Beta-Glucans in standaridized allergen extracts. J Endotoxin Res. 12(4):241-245 (2006).
- Finlay WJ, deVore NC, Dobrovolskaia EN, Gam A, Goodyear CS, Slater JE. Exploiting the avian immunoglobulin system to simplify the generation of recombinant antibodies to allergenic proteins. Clin Exp Allergy. 35(8):1040-1048 (2005).
- Dobrovolskaia E, Gam A, Slater JE.Competition enzyme-linked immunosorbant assay (ELISA) can be a sensitive method for the specific detection of small quantities of allergen in a complex mixture. Clin Exp Allergy. 36(4):525-530 (2006).
- Epstein SL. Prior H1N1 influenza infection and susceptibility of Cleveland Family Study participants during the H2N2 pandemic of 1957: An experiment of nature. Journal of Infectious Diseases. 193:49-53 (2006).
- Epstein SL, Kong WP, Misplon JA, Lo CY, Tumpey TM, Xu L, Nabel GJ. Protection against multiple influenza A subtypes by vaccination with highly conserved nucleoprotein. Vaccine. 23:5404-5410 (2005).
- Han J, Farnsworth RL, Tiwari JL, Tian J, Lee H, Ikonomi P, Byrnes AP, Goodman JL, Puri RK. Quality prediction of cell substrate using gene expression profi ling. Genomics. 87:552-559 (2006).
- Peden K, Sheng L, Pal A, Lewis A. Biological activity of residual cell-substrate DNA. Dev Biol. 123:45-53 (2006).
- Tomioka K, Peredelchuk M, Zhu X, Arena R, Volokhov D, Selvapandiyan A, Stabler K, Mellquist-Riemenschneider J, Chizhikov V, Kaplan G, Nakhasi H, Duncan R. A multiplex polymerase chain reaction microarray assay to detect bioterror pathogens in blood. J Mol Diagn. 7(4):486-494 (2005).
- Wang H, May J, Del Grosso A. Determination of benzethonium chloride in anthrax vaccine adsorbed by HPLC. Biologicals. 34(4):257-63. (Epub Feb 18, 2006).
- Herrmann JE, Wang S, Zhang C, Panchal RG, Bavari S, Lyons CR, Lovchik JA, Golding B, Shiloach J, Lu S. Passive immunotherapy of Bacillus anthracis pulmonary infection in mice with antisera produced by DNA immunization. Vaccine. 24(31-32):5872-5880 (2006).
- Klinman DM. CpG oligonucleotides accelerate and boost the immune response elicited by AVA, the licensed anthrax vaccine. Expert Rev Vaccines. 5(3):365-369 (2006).
- Biological Products; Bacterial Vaccines and Toxoids; Implementation of Effi cacy Review; Anthrax Vaccine Adsorbed; Final Order. Federal Register 2005; 70 (242):75180-75198.
- Bryant-Genevier MG, Sommer S, McMahon AW, Ball R, Braun MM. Correlates of public health workforce acceptance of smallpox immunization in Virginia. Public Health Nursing. 23:339-346 (2006).
- Heraud JM, Edghill-Smith Y, Ayala V, Kalisz I, Parrino J, Kalyanaraman VS, Manischewitz J, King LR, Hryniewicz A, Trindade CJ, Hassett M, Tsai WP, Venzon D, Nalca A, Vaccari M, Silvera P, Bray M, Graham BS, Golding H, Hooper JW, Franchini G. Subunit recombinant vaccine protects against monkeypox. J Immunol. 177(4):2552-2564 (2006).
- Bryant-Genevier M, O'Connell K, Ball R, Braun MM, McMahon A. Passive surveillance for generalized vaccinia in the United States using the Vaccine Adverse Event Reporting System (VAERS). Vaccine. 24(17):3632-3635 (2006).
- Jones-Trower A, Garcia A, Meseda CA, He Y, Weiss C, Kumar A, Weir JP, Merchlinsky M. Identifi cation and preliminary characterization of vaccinia virus (Dryvax) antigens recognized by vaccinia immune globulin. Virology. 343(1): 128-140 (2005).
- Srinivasan K, Wood O, Akolkar P, Lee S, Hewlett I. Absence of detectable viremia in the plasma of recent Smallpox vaccinees. Transfusion. 46(9):1589-92 (2006).
- Rios M, Zhang MJ, Srinivasan K, Daniel S, Hewlett I, Dayton A. Human primary monocyte-derived macrophages (MDM) support West Nile virus (WNV) replication in vitro. Transfusion. 46:659-667 (2006).
- Rios M, Zhang MJ, Grinev A, Srinivasan K, Daniel S, Wood O, Hewlett IK, Dayton AI. Monocytes-macrophages are potential target in human infection with West Nile virus through blood transfusion. Transfusion. 46(4):659-667 (2006).
- Haddad SA, Lichtiger B, Klein HG. In vivo effi cacy of shipped HLA-matched platelets. Transfusion. 46(8):1306-1310 (2006).
- Burwen D, La Voie L, Braun MM, Houck P, Ball R. Evaluating adverse events after vaccination in the Medicare population. Pharmacoepidemiology and Drug Safety. 15(Supplement 1):S79-80 (2006).
- Freedman SB, Reed J, Burwen DR, Wise RP, Weiss A, Ball R. Transient bulging fontanelle after vaccination: Case report and review of the Vaccine Adverse Event Reporting System (VAERS). Journal of Pediatrics. 147:640-644 (2005).
- Woo EJ, Miller NB, Ball R. Adverse events after hepatitis A B combination vaccine. Vaccine. 24:2685-2691 (2006).
- Epstein SL, Kong WP, Misplon JA, Lo CY, Tumpey TM, Xu L, Nabel GJ. Protection against multiple influenza A subtypes by vaccination with highly conserved nucleoprotein. Vaccine. 23(46-47):5404-5410 (2005).
- Kioi M et al. Interleukin-13 receptor alpha2 chain: a potential biomarker and molecular target for ovarian cancer therapy. Cancer. 107(6):1407-1418 (2006).
- Mpanju OM, Towner JS, Dover JE, Nichol ST, Wilson CA. Identifi cation of two amino acid residues on Ebola virus glycoprotein 1 critical for cell entry. Virus Res. 121(2):205-214 (2006).
- Rubin S, Mauldin J, Chumakov K, Vanderzanden J, Iskow R, Carbone K. Serological and phylogenetic evidence of monotypic immune responses to different mumps virus strains. Vaccine. 24(14):2882-2888 (2006).
- Khurana S, Needham J, Mathieson B, Rodriguez-Chavez IR, Catanzaro AT, Bailer RT, Kim J, Polonis V, Cooper DA, Guerin J, Peterson ML, Gurwith M, Nguyen N, Graham BS, Golding H. Human Immunodefi ciency Virus (HIV) vaccine trials: a novel assay for differential diagnosis of HIV infections in the face of vaccine-generated antibodies. J Virol. 80(5):2092-2099 (2006).
- Steele AD, Warfel JM, D'Agnillo F. Anthrax lethal toxin enhances cytokine-induced VCAM-1 expression on human endothelial cells. Biochem Biophys Res Commun. 337(4):1249-1256 (2005).
- Maximova OA, Taffs RE, Pomeroy KL, Piccardo P, Asher DM. Computerized morphometric analysis of pathological prion protein deposition in scrapie-infected hamster brain. J Histochem Cytochem. 54(1):97-107 (2006).
- Farshid M, Taffs RE, Scott D, Asher DM, Brorson K. The clearance of viruses and transmissible spongiform encephalopathy agents from biologicals. Curr Opin Biotechnol. 16(5):561-567 (2005).
Appendix B (CBER Exhibit Program - FY 2006)
CBER Exhibit Program - FY2006
Meeting |
Dates |
|---|---|
American Association of Blood Banks Seattle, WA |
October 14-19, 2005 |
Maryland PTA Fall Meeting Hunt Valley , MD |
November 11, 2005 |
NBC4 Health and Fitness Expo Washington, DC |
January 14-15, 2006 |
BioChem Defense & Pandemic Vaccine & Therapeutics Washington, DC |
April 24-26, 2006 |
FDA Science Forum Washington, DC |
April 18-19, 2006 |
National Foundation for Infectious Diseases Conference on Vaccine Research Baltimore, MD |
May 8-10, 2006 |
Drug Information Association Philadelphia, PA |
June 17-21, 2006 |
American Association of Tissue Banks Annual Meeting San Diego, CA |
September 8-12, 2006 |
International Society of Cellular Therapy Bethesda, MD |
September 25-27, 2006 |
APPENDIX C (CBER Major Approvals - FY 2006)
Biologic License Applications
Tradename/ Proper Name |
Indication for Use |
Manufacturer |
|---|---|---|
ABBOTT PRISM HBcore Hepatitis B Virus Core Antigen |
Qualitative detection of total antibody to hepatitis B core antigen (anti-HBc) in human serum and plasma; screening test for blood and plasma to prevent transmission of hepatitis B virus (HBV) to recipients of blood and blood components. |
Abbott Laboratories |
ABBOTT PRISM HBsAg; ABBOTT PRISM HBsAg Confirmatory |
ABBOTT PRISM HBsAg and ABBOTT PRISM HBsAg Confi rmatory assay, in-vitro microparticle chemiluminescent immunoassays (ChLIA) for the qualitative detection of HBsAg in human serum or plasma. |
Abbott Laboratories |
GARDASIL |
Vaccination in females 9 to 26 years of age for prevention of the following diseases caused by Human Papillomavirus (HPV) Types 6, 11, 16, and 18:
|
Merck & Co., Inc. |
Biologics License Applications (Continued)
Tradename/ Proper Name |
Indication for Use |
Manufacturer |
|---|---|---|
HepaGam B Hepatitis B Immune Globulin (Human) |
Treatment of acute exposure to blood containing HBsAg, perinatal exposure of infants born to HBsAg-positive mothers, sexual exposure to HBsAg-positive persons and household exposure to persons with acute HBV infection. |
Cangene Corp. 104 |
Procleix WNV Assay |
Qualitative detection of West Nile virus (WNV) RNA in plasma specimens from individual human donors, including volunteer donors of whole blood and blood components, and other living donors; testing plasma specimens to screen organ donors when specimens are obtained while the donor's heart is still beating, and testing blood specimens to screen cadaveric (non-heart-beating) donors. It is not intended for use on cord blood specimens. |
Gen-Probe Inc. |
RotaTeq Rotavirus Vaccine, Live, Oral, Pentavalent |
Prevention of rotavirus gastroenteritis in infants and children caused by the serotypes G1, G2, G3, and G4 when administered as a 3-dose series to infants between the ages of 6 to 32 weeks. |
Merck & Co., Inc. |
Vivaglobin Immune Globulin Subcutaneous (Human) |
Treatment of patients with primary immune defi ciency (PID). |
ZLB Behring, L.L.C. |
Zostavax Zoster Vaccine, Live, (Oka/Merck ) |
Prevention of herpes zoster (shingles) in individuals 60 years of age and older when administered as a single-dose. |
Merck & Co., Inc. |
Biologics License Supplements
(for New Indications, New Routes of Administration, New Dosage Forms, Improved Safety)
Tradename/ Proper Name |
Indication for Use |
Manufacturer |
|---|---|---|
Dryvax |
Package Insert Revision: |
Wyeth Pharmaceuticals, Inc. |
Fluarix |
New Formulation: 2006-2007 United States formulation |
GlaxoSmithKline Biologicals |
FluMist |
New Formulation: 2006-2007 United States formulation |
MedImmune Vaccines, Inc. |
Fluvirin |
New Formulation: 2006-2007 United States formulation |
Chiron Vaccines Ltd. |
Havrix |
Expanded Indication: Age indication lowered from two years to 12 months. |
GlaxoSmithKline Biologicals |
Pediarix |
Package Insert Revision: safety and immunogenicity of a three dose series of Pediarix when administered with other recommended US-licensed vaccines including Prevnar. |
GlaxoSmithKline Biologicals |
Tisseel VH Kit |
New formulation: To include the incorporation of a second, independent virus inactivation step (solvent/detergent treatment); modifi cations to the manufacturing process and manufacturing locations within the Vienna facilities; and the introduction of a frozen liquid presentation of the product. |
Baxter Healthcare Corp. |
New Drug Applications
Tradename/ Proper Name |
Indication for Use |
Manufacturer |
|---|---|---|
MacoProductions S.A.S. CPD/AS-1: MacoPharma Leucoflex MTL1 Leukoreduction System for Blood |
For pre-storage leukocyte reduction of whole blood initiated between 4 and 7 hours after collection if whole blood is stored at ambient temperature, or between 4 and 8 hours of storage at 1°C to 6°C. |
MacoProductions S.A.S. |
Device Applications
Tradename/ Proper Name |
Indication for Use |
Manufacturer |
|---|---|---|
ADVIA Centaur HIV 1/O/2 Enhanced ReadyPack Reagents |
For the qualitative determination of antibodies to the human immunodeficiency virus type 1, including group O, and/or type 2 in serum or plasma using the ADVIA Centaur System. |
Bayer HealthCare |
Chembio HIV Reactive/ Nonreactive Controls SURE CHECK HIV 1/2 ASSAY |
For the detection of antibodies to HIV-1 and HIV-2 in finger stick whole blood, venous whole blood, and serum or plasma specimens. |
Chembio Diagnostic Systems, Inc. 3661 Horseblock Road |
Chembio HIV 1/2 STAT-PAK ASSAY |
For the detection of antibodies to HIV-1 and HIV-2 in finger stick whole blood, venous whole blood, and serum or plasma specimens. |
Chembio Diagnostic Systems, Inc. 3661 Horseblock Road |
Appendix D (Rulemaking and Guidance Documents - FY 2006)
Rulemaking and Guidance Documents for FY2006
I. RULEMAKINGS
A. The following proposed and final rules and final orders were issued by CBER and published in the Federal Register in FY 2006:
- Medical Device Regulations; Addresses; Technical Amendment; Final
Rule-7/25/2006 - Blood Vessels Recovered With Organs and Intended for Use in Organ Transplantation; Direct Final Rule and Companion Document to Direct Final Rule; Proposed Rule--5/12/2006
- Distribution of Blood Derivatives by Registered Blood Establishments that Qualify as Health Care Entities; Prescription Drug Marketing Act of 1987; Prescription Drug Amendments of 1992; Policies, Requirements and Administrative Procedures; Proposed Rule--2/1/2006
- Biological Products; Bacterial Vaccines and Toxoids; Implementation of Efficacy Review; Final Rule and Final Order--12/15/2005
- Biological Products; Bacterial Vaccines and Toxoids; Implementation of Efficacy Review; Anthrax Vaccine Adsorbed; Final Order--12/15/2005
- Revocation of Status of Specific Products; Group A Streptococcus; Direct Final Rule and Companion Document to Direct Final Rule; Proposed Rule--12/2/2005
B. CBER was involved in the clearance of the following published proposed and final rules for which other FDA Centers/Offices were the lead:
- Requirements for Foreign and Domestic Establishment Registration and Listing for Human Drugs, Including Drugs that are Regulated Under a Biologics License Application, and Animal Drugs; Proposed Rule--8/29/2006
- Requirements on Content and Format of Labeling for Human Prescription Drug and Biological Products; Final Rule--1/18/2006
- Current Good Manufacturing Practice Regulation and Investigational New Drugs; Direct Final Rule and Companion Document to Direct Final Rule; Proposed Rule--1/17/2006
- Investigational New Drugs: Export Requirements for Unapproved New Drug Products; Final Rule--11/23/2005
II. GUIDANCE DOCUMENTS
A. The following guidance documents were issued by CBER and posted and/or
published in FY2006:
- Guidance for Industry: Quality Systems Approach to Pharmaceutical CGMP Regulations - 9/29/2006
- International Conference on Harmonisation (ICH); Guidance for Industry: E5
- Ethnic Factors in the Acceptability of Foreign Clinical Data - Questions and Answers -- 9/28/2006
- Draft Guidance for Industry: Characterization and Qualification of Cell Substrates and Other Biological Starting Materials Used in the Production of Viral Vaccines for the Prevention and Treatment of Infectious Diseases - 9/28/2006
- Guidance for Industry: Recognition and Use of a Standard for Uniform Blood and Blood Component Container Labels - 9/22/2006
- United States Industry Consensus Standard for the Uniform Labeling of Blood and Blood Components Using ISBT 128 - 9/22/2006
- Guidance for Industry and FDA Staff: Minimal Manipulation of Structural Tissue Jurisdictional Update - 9/20/2006
- Draft Guidance for Industry: Drug Interaction Studies - Study Design, Data
Analysis, and Implications for Dosing and Labeling - 9/11/2006 - Guidance for Industry: Compliance with 21 CFR Part 1271.150(c)(1) -
Manufacturing Arrangements - 9/8/2006 - Guidance for Industry: Implementing a Collection Program for Source Plasma Containing Disease-Associated and Other Immunoglobulin G (IgG) Antibodies -8/8/2006
- Draft Guidance for Industry: Amendment (Donor Deferral for Transfusion in France Since 1980) to "Guidance for Industry: Revised Preventive Measures to Reduce the Possible Risk of Transmission of Creutzfeldt-Jakob Disease (CJD) and Variant Creutzfeldt-Jakob Disease (vCJD) by Blood and Blood Products" - 8/8/2006
- Guidance for Industry: Providing Regulatory Submissions to the Center for
Biologics Evaluation and Research (CBER) in Electronic Format - Lot Release Protocols -/12/2006 - Guidance for Industry: Development of Preventive HIV Vaccines for Use in
Pediatric Populations - 5/4/2006 - Draft Guidance for Industry: Informed Consent Recommendations for Source Plasma Donors Participating in Plasmapheresis and Immunization Programs - 4/25/2006
- Guidance for Clinical Trial Sponsors: Establishment and Operation of Clinical Trial Data Monitoring Committees - 3/27/2006
- Draft Guidance for Industry: Clinical Data Needed to Support the Licensure of Pandemic Influenza Vaccines - 3/2/2006
- Draft Guidance for Industry: Clinical Data Needed to Support the Licensure of Trivalent Inactivated Influenza Vaccines - 3/2/2006
- Guidance for Industry: Considerations for Developmental Toxicity Studies
for Preventive and Therapeutic Vaccines for Infectious Disease Indications - 2/13/2006 - Draft Guidance for Industry: Safety, Efficacy, and Pharmacokinetic Studies to Support Marketing of Immune Globulin Intravenous (Human) as Replacement Therapy for Primary Humoral Immunodefi ciency - 11/30/2006
- Guidance for Industry: MedWatch Form FDA 3500A: Mandatory Reporting of Adverse Reactions Related to Human Cells, Tissues, and Cellular and Tissue- Based Products (HCT/Ps) - 11/30/2006
B. CBER was involved in the clearance of the following published draft and final guidances for which other FDA Centers/Offices were the lead:
- Draft Guidance for Industry: Drug Interaction Studies - Study Design, Data
Analysis, and Implications for Dosing and Labeling - 9/11/2006 - Draft Guidance for Institutional Review Boards, Clinical Investigators, and
Sponsors: Exception from Informed Consent Requirements for Emergency
Research - 9/7/2006 - Annual Guidance Agenda - 9/6/2006
- International Conference on Harmonisation (ICH); Draft Guidance: Q4B Regulatory Acceptance of Analytical Procedures and/or Acceptance Criteria (RAAPAC) - 8/7/2006
- International Conference on Harmonisation (ICH); Guidance for Industry: Q3B(R2) Impurities in New Drug Products - 7/31/2006
- Guidance for Industry: Chronic Cutaneous Ulcer and Burn Wounds - Developing Products for Treatment - 6/1/2006
- International Conference on Harmonisation (ICH); Guidance for Industry: Q9 Quality Risk Management - 6/1/2006
- International Conference on Harmonisation (ICH); Guidance for Industry: Q8 Pharmaceutical Development - 5/19/2006
- Guidance for Industry and FDA Staff: Real-Time Premarket Approval Application (PMA) Supplements - 4/28/2006
- Guidance for Industry: Bar Code Label Requirements - Questions and Answers - 4/27/2006
- Guidance for Sponsors, Institutional Review Boards, Clinical Investigators and FDA Staff: Guidance on Informed Consent for In Vitro Diagnostic Device Studies Using Leftover Human Specimens that are Not Individually Identifiable - 4/25/2006
- Guidance for Industry: Providing Regulatory Submissions in Electronic Format -- Human Pharmaceutical Product Applications and Related Submissions Using the eCTD Specifications - 4/19/2006
- International Conference on Harmonisation (ICH); Guidance for Industry: S8 Immunotoxicity Studies for Human Pharmaceuticals - 4/12/2006
- Annual Comprehensive List of Guidance Documents at the Food and Drug Administration - 3/28/2005
- Guidance for Industry: Using a Centralized IRB Review Process in Multicenter Clinical Trials - 3/15/2006
- Guidance for Industry: Reports on the Status of Postmarketing Study Commitments - Implementation of Section 130 of the Food and Drug Administration Modernization Act of 1997 - 2/15/2006
- Draft Guidance for Industry: Patient-Reported Outcome Measures: Use in Medical Product Development to Support Labeling Claims - 2/2/2006
- Guidance for Industry: Adverse Reactions Section of Labeling for Human Prescription Drug and Biological Products - Content and Format - 1/18/2006
- Guidance for Industry: Clinical Studies Section of Labeling for Human Prescription Drug and Biological Products - Content and Format - 1/18/2006
- Draft Guidance for Industry: Warnings and Precautions, Contraindications, and Boxed Warning Sections of Labeling for Human Prescription Drug and Biological Products - Content and Format - 1/18/2006
- Draft Guidance for Industry: Labeling for Human Prescription Drug and Biological Products - Implementing the New Content and Format Requirements - 1/18/2006
- Draft Guidance for Industry: INDs - Approaches to Complying with CGMP During Phase 1 - 1/12/2006
- Guidance for Industry: Formal Dispute Resolution: Scientific and Technical Issues Related to Pharmaceutical CGMP - 1/11/2006
- Guidance for Industry: Fast Track Drug Development Programs - Designation, Development, and Application Review - 1/11/2006
- Information Sheet Guidance for Sponsors, Clinical Investigators, and IRBs: Waiver of IRB Requirements for Drug and Biological Product Studies - 1/2006
- International Conference on Harmonisation (ICH); Guidance for Industry: E14 Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs - 10/19/2005
- International Conference on Harmonisation (ICH); Guidance for Industry: S7B Nonclinical Evaluation of the Potential for Delayed Ventricular Repolarization (QT Interval Prolongation) by Human Pharmaceuticals - 10/19/2005
- International Conference on Harmonisation (ICH); Guidance for Industry: Granularity Document Annex to M4: Organization of the CTD - 10/18/2006
Appendix E (Advisory Committee Meetings - FY 2006)
Allergy Products Advisory Committee
September 13, 2006
- Open Discussion Topics: FDA Proposed Strategy for the Reclassification of Category IIIA Allergenic Products; Research Update: Laboratory of Immunochemistry, Division of Bacterial, Parasitic and Allergenic Products
- Closed Committee Discussion Topics: Individual research programs in the Division of Bacterial, Allergenic and Parasitic Products, Office of Vaccines Research and Review
Blood Products Advisory Committee
November 3-4, 2005
- Updates: West Nile Virus; Draft Guidance on NAT for HIV-1 and HCV; Summary of the TSEAC meeting held Oct. 31, 2005; Summary of DHHS Advisory Committee on Blood Safety and Availability; Re-entry of Donors Deferred Based on anti-HBc TestResults
- Open Committee Discussion: Approaches to Over-the-Counter (OTC) Home-Use HIV Test Kits
- Updates: Informational presentation on Serious Adverse Events
- Following Falsely Elevated Glucose Measurements Resulting from Administration of an IGIV Product Containing Maltose
- Open Committee Discussion: Heterogeneity of Commercial Alpha-1-Proteinase Inhibitor (Human) Products - Implications for Longer-Term Safety and Efficacy
March 9-10, 2006
- Committee Updates: Meeting Summary of DHHS Advisory Committee on Blood Safety and Availability; Current Consideration for Blood Donor Screening for West Nile Virus; Classification of Transfusion Recipient ID Systems; Workshop Summary - Behavior-Based Donor Deferrals in the NAT ERA
- Open Discussion Topics: Rapid Tests for Detection of Bacterial Contamination of Platelets
- Committee Update: Current Considerations on Implementation of New Recommendations for Donor Eligibility
- Open Discussion Topics: Public Comments on "Guidance for Industry and FDA Review Staff : Collection of Platelets by Automated Methods; Proposed Studies to Support the Approval of Over-the-Counter Home-Use HIV Test Kits; Summary of the Office of Blood Research and Review Site Visit (July 22, 2005); Review of Research Programs Site Visit of the Division of Hematology Laboratories, OBRR (Oct. 6, 2005)
- Closed Committee Discussion Topics: Office of Blood Research and Review Site Visit and Individual research programs in the Division of Hematology Laboratories
July 13, 2006
- Committee Updates: Summary of May 9-10, 2006 Meeting of the DHHS Advisory Committee on Blood Safety and Availability; Summary of July 12, 2006 FDA Workshop of Testing for Malarial Infections in Blood Donors; Committee Report on the Office of Blood Research and Review Site Visit; West Nile Virus Update
- Open Committee Discussions: FDA Review of Nabi Biopharmaceuticals' Hepatutis B IGIV for Prevention of Recurrent HBV Disease After Orthotopic Liver Transplantation; Review of the Research Programs in the Laboratory of Bacterial, Parasitic and Unconventional Agents, Division of Emerging and Transfusion Transmitted Diseases
- Closed Committee Discussion Topics: Individual research programs Division of Emerging and Transfusion Transmitted Diseases, Laboratory of Bacterial, Parasitic and Unconventional Agents
Cellular, Tissue, and Gene Therapies Advisory Committee
February 9-10, 2006
- Open Discussion Topics: Potency Measurements for Cell and Gene
Therapy Products; National Toxicology Program, Proposed Study on Retroviral Vector-Mediated Insertional Mutagenisis; Overview - Research Program, Office of Cellular, Tissue and Gene Therapies - Closed Committee Discussion Topics: Report of the Office of
Cellular, Tissue and Gene Therapies Research Program
Transmissible Spongiform Encephalopathies Advisory Committee
October 31, 2005
- Open Committee Discussion: Progress Report on FDA's Risk Assessment for Potential Exposure to Variant Creutzfeldt-Jakob Disease in Human Plasma-Derived Antihemophilic Factor (FVIII) Products; Labeling Claims for Filters Intended to Remove TSE Infectivity; from Blood Components
- Committee Updates: US and Worldwide BSE Status; Scientific Issues in Evaluating Products Intended to Decontaminate Surgical Instruments Exposed to TSE Agents; Discussions of a Recent FDA Device Panel
September 18-19, 2006
- Committee Updates: US and Worldwide BSE; vCJD Epidemiology and Transfusion-Transmission; Draft Guidance for Industry Amendment; Critical Factors Influencing Prion Decontamination Using Sodium Hydroxide - PPTA Collaborative Study; Human Prions: Clearance and Plasma Lipoproteins
- Open Committee Discussion: Experimental Clearance of Transmissible Spongiform Encephalopathy Infectivity in Plasma-derived FVIII Products;
- Committee Updates: Status of FDA's Initiative on Communication of the Potential Exposure to vCJD risk From an Investigational Product, Plasma Derived Factor XI That Was Manufactured From UK Donor Plasma; Summary of World Health Organization Consultation on Distribution of Infectivity in Tissues of Animals and Humans with TSE
- Open Committee Discussion: Possible Criteria for Approval of a Donor Screening Test for vCJD
Vaccines and Related Biological Products Advisory Committee
November 16-17, 2005
- Open Committee Discussion: Use of MDCK Cells for Manufacture of Inactivated Influenza Virus Vaccines; Developing New Pneumococcal Vaccines for U.S. Licensure for Adults
- Closed Committee Discussion
December 14-15, 2005
- Open Committee Discussion: Safety and Efficacy of Rotavirus
Vaccine Manufactured by Merck; Safety and Efficacy of ZOSTAVAX® (Zoster Vaccine Live [Oka/Merck]) Manufactured by Merck
February 17, 2006
- Open Committee Discussion: Strain Selection for InfluenzaVirus for the 2006-2007 Season
May 18, 2006
- Open Committee Discussion: Safety and Efficacy of GARDASIL® (Human Papillomavirus Types 6, 11, 16, 18) Recombinant Vaccines by Merck
Appendix F (Organizational Chart)