|
| |
||
| |
|||||||||||||||||
|
|||||||||||||||||
|
|||||||||||||||||
|
EID Home | Ahead of Print | Past Issues | EID Search | Contact Us | Announcements | Suggested Citation | Submit Manuscript
|
|
Research Molecular Analysis of Plasmodium ovale VariantsThin Thida Win,* Amadu Jalloh,* Indah Setyawati Tantular,† Takafumi
Tsuboi,‡ Marcelo Urbano Ferreira,§ Masatsugu Kimura,¶ and Fumihiko Kawamoto*
The geographic range of the human malaria parasite Plasmodium ovale has been thought to be mostly limited to tropical Africa, the Middle East, Papua New Guinea, and Irian Jaya in Indonesia; it has rarely been described in other countries of Southeast Asia. More recently, however, with the aid of polymerase chain reaction (PCR)-based species identification and improved microscopic techniques, P. ovale infections have been frequently reported in Southeast Asia (1,2). P. ovale may represent an emerging cause of benign and relapsing tertian malaria in this region or, alternatively, may have been overlooked in previous surveys based on classic microscopy techniques (1). The widespread distribution of P. ovale in Southeast Asia affects the choice of appropriate drugs for malaria chemoprophylaxis in travelers, since most currently used regimens are not effective against the dormant liver stages of P. ovale and P. vivax, which may cause relapses several months after the primary infection (3). During our previous molecular studies of P. ovale in southern Vietnam (4), we found two field isolates whose partial sequences at the block 9 region (5) of the small subunit ribosomal RNA (SSUrRNA) genes had a deletion of 2 nt (G-G) and a substitution of 1 nt (C to T), when compared with the classic type, the Nigerian I/CDC strain (6). These polymorphisms had practical implications, since they occurred in the target of a diagnostic oligonucleotide probe used by the commercially available microtiter-plate hybridization (MPH) method for malaria diagnosis (4). Soon after, the same sequence variation was reported in three cases imported from Africa into Japan (7); all patients had single infections with the variant P. ovale. Later, variant-type sequences were found in the Cameroon (8) and MAL/MAI isolates (L.K. Basco, unpub. data), as well as in isolates from other Southeast Asian countries such as Thailand, Laos, Myanmar, and Indonesia (9–12). Four features of sequence variation in P. ovale soon became clear: 1) both classic and variant-type parasites occurred in sympatry (i.e., they co-occurred in the same disease-endemic areas); 2) parasites with variant-type sequences did not differ morphologically from classic parasites; 3) variant-type parasites were present in both Asia and Africa; and 4) parasites with variant-type sequences tended to produce higher parasitemia levels and higher proportions of single-species infection, when compared with classic P. ovale infections acquired in the same region (2,11). In contrast with P. falciparum and P. vivax, little is known about the patterns of genetic diversity in field isolates of P. ovale. So far, full sequences of the SSUrRNA gene have been analyzed for only three isolates, the Nigerian I/CDC strain (6) and two African isolates, Cameroon and MAL/MAI; partial sequences are also available only for four isolates, the London School of Hygiene and Tropical Medicine strain (LS train) and the Nigerian I/CDC strain (13), and two isolates from Papua New Guinea (14) and Ghana (C. Severini et al., unpub. data). The cysteine protease gene was sequenced only for the Nigerian I/CDC (15), whereas the Harding strain is the only source of available sequence for the cytochrome b (cyt b) gene (16). More recently, two types of sequences have been characterized for the ookinete surface protein genes, Pos 25, Pos 28-1, and Pos 28-2 in P. ovale isolates from Thailand (17,18). They correspond to the two types of SSUrRNA genes, Nigerian I/CDC and LS, which suggests that two sequence types might represent distinct variants or subspecies (13,18). We have obtained sequence data of the SSUrRNA, cysteine protease, ookinete surface protein, and cyt b genes of P. ovale isolates from Myanmar and Indonesia and compared our data with GenBank-available sequences. Our analyses of both nuclear and mitochondrial genes provide further support to the division of P. ovale into at least two types. Materials and MethodsField P. ovale IsolatesAll isolates were obtained during our recent field surveys in Myanmar and Indonesia (2,11,12). For molecular analysis of the variant and classic types, patients with single infections were selected. The variant isolates we analyzed were ST243 (Rakhine State) and MC53 (Tanintharyi Division), both from Myanmar, and M474 (Flores Island, eastern Indonesia). The three classic isolates of M3 (Shan State), M4 (Bago Division), and T134 (Mon State) were collected from Myanmar. Isolation of Parasite DNA and Confirmation of P. ovale by Sequence AnalysisParasite DNA templates were isolated from blood by using a DNA isolation Kit (High Pure PCR Template Preparation Kit, Boehringer Mannheim, Germany). Then the target sequences at the block 9 region used for PCR-based diagnosis were further analyzed to confirm the presence of the variant- or classic-type in P. ovale–positive samples. Amplified DNA products using the P1F-Up and specific reverse (PoR2) primers (Table 1) underwent direct sequencing, whereas the first PCR products were cloned into the pCR II plasmid from a TA Cloning Kit (Invitrogen, San Diego, CA). The target fragments of 12 positive clones from each sample were sequenced by using Big Dye Terminator sequencing kit on an ABI 310 sequencer (PE Applied Biosystems, Foster City, CA. Analysis of SSUrRNA, Cysteine Protease, and cyt b GenesPrimer sets used were shown in Table 1. For analysis of the SSUrRNA gene, PCR amplification was performed by using AmpliTaq Gold polymerase (PE Applied Biosystems) at 96°C for 10 min, 36 cycles at 94°C for 30 sec, 55°C for 1 min, and 72°C for 2 min, followed by one cycle at 72°C for 10 min. For analysis of the cysteine protease and cyt b genes, PCR conditions were slightly modified from the original methods (15,16). The conditions used were one cycle at 96°C for 10 min, 36 cycles at 94°C for 30 s, 50°C for 1 min, and 72°C for 90 s, followed by one cycle at 72°C for 10 min. The amplified PCR products were cloned into the pCR II vector. Plasmid DNA was purified from the positive colonies and sequenced in both directions by using the primers described in Table 1 in combination with M13 primers. Sequencing was performed with an ABI 377 sequencer. Any ambiguity and putative polymorphism was checked by additional amplification and sequencing. Sequences obtained were compared with those reported in databases. Gene sequences used for the SSUrRNA genes were clone 9 and 26 of the Nigerian I/CDC strain (6), isolates from Cameroon (8), MAL/MAI (X99790), Papua New Guinea (14), Ghana (AJ250701), and two strains of the Nigerian I/CDC and the London School of Hygiene and Tropical Medicine (13). For the cysteine protease, P. ovale Nigerian I/CDC, P. malariae WR314, P. cynomolgi, P. reichenowi (15), P. vivax Salvador-1 (19), and P. falciparum (20) were used. For the cyt b. P. ovale Harding strain, P. malariae Uganda-1, P. falciparum Kenya, Santa Lucia, Malaysian-4, 3D7, P. simiovale, P. knowlesi, P. cynomolgi (16), P. falciparum Malay Camp (21) and C10 (22), P. reichenowi, P. falciparum NF54, K1, T9/96, 7G8 (23), Indian isolate 317 (24), P. vivax Salvador-I (25), and Indian PH 10 (26) were retrieved as well as P. berghei (27) and P. yoelii (28). Dendrograms were obtained with PHYLIP (Version 3.5c, University of Washington, Seattle, WA) by using the neighbor-joining method with a Kimura’s two-parameter distance and the maximum likelihood method. Sequence Analysis of Pos 25, Pos 28-1, and Pos 28-2 GenesThe procedures for first and nested PCR amplifications with primers (Table 1) were described previously (17,18). Nucleotide sequences were determined by direct sequencing with nested PCR products. Then, sequences obtained were compared with those reported previously (AB051631-3, AB074973-6). ResultsSequence Analysis of the Full SSUrRNA Gene
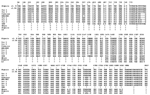
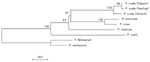
Three different sequences were obtained for the SSUrRNA gene from each variant-type isolate, while two different sequences were detected from each classic-type isolate (Figure 1). However, whether all of them were A (asexual)-type genes or sequences included S (sexual)- or O (ookinete/oocyst)-type (5,29,30) genes was unknown. Hereafter, these sequences are referred to as Pov 1–3 for the variant-type and as Poc 1–2 for the classic-type. The differences among Pov 1–3 were seen at the 5´ half (Figure 1). When compared with the four complete sequences in GenBank, the Cameroon and MAL/MAI isolates were grouped as variant-type (>99% identity with Pov 1–3 and <97% with Poc 1–2). Both African isolates also shared the same mutation at the block 9 region (nucleotide positions of 1158–1160). Particularly, the sequence found in Cameroon isolate resembled that of Pov 1 (only 4-bp difference). The alignment of four partial sequences showed that, despite their same origin, the partial sequence of the Nigerian I/CDC (13) also showed 9-bp and 5-bp differences from those of clones 9 and 26, respectively. Among these isolates, the LS strain possessed the same sequence as the 3´ half of Pov 1–3, and thus it was grouped as variant type (<96% identity with Poc 1–2). The sequence of the Papua New Guinea isolate was more similar to that of clone 9 of the Nigerian I/CDC (>98.8% identity with Poc 1 >and <97% with Pov 1–3) than to that of clone 26 or Poc 2 (98.2%–98.4% identity). The Ghana isolate was also grouped as classic-type (>97% identity with the Nigerian I/CDC or Poc 1–2 and <92% with Pov 1–3). These results suggest that the Papua New Guinea, Ghana, and our classic isolates are members of the classic- (Nigerian I/CDC-) type group, whereas the Cameroon and MAL/MAI isolates, as well as our variant isolates, are all members of the variant- (LS-) type group. Sequence Analysis of the Cysteine Protease and the Ookinete Surface Protein GenesThe analysis of 531 bp of the cysteine protease genes, when compared with the reported sequence of the Nigerian I/CDC, showed that variant isolates differed at 19 bp (3.6%) with eight nonsilent mutations and that classic-type isolates had an almost identical sequence, except for a single base at position 700 (nonsilent substitution from Pro to Ala) (Table 2). Because the same substitution is also seen in the variant P. ovale, P. falciparum, P. vivax, P. malariae, P. reichenowi, and P. cynomolgi (data not shown), this nucleotide may have been misread in the sequence of the Nigerian I/CDC strain; if so, sequences of classic-type isolates are identical to those of the Nigerian I/CDC strain. At the amino acid level, the variant isolates showed 96.0% sequence identity with the classic isolates. Tachibana et al. (17) have analyzed the ookinete surface protein genes in Thai isolates and reported that two (Nigerian I/CDC and LS) types of P. ovale, defined by the SSUrRNA genes, have distinct sequences. Our nearly complete sequences of Pos 25, Pos 28-1, and Pos 28-2 in variant isolates were identical to those found in LS-type Thai isolates, while sequences in the classic isolates were identical to those of the Nigerian I/CDC-type (data not shown). Sequence Analysis of the cyt b GeneThe analysis of 1035 bp of the cyt b genes showed that variant- and classic-type P. ovale isolates differed from each other at 12 bp, with one nonsilent substitution (Table 3). Sequences of variant isolates differed from those reported for the Harding strain at 15 bp (1.4%), with two nonsilent substitutions. The SSUrRNA gene of the Harding strain was not reported yet, and whether this strain is of the classic or variant type is not known. From its cyt b sequence, it was expected that this strain may belong to the classic group, despite the differences between the respective sequences (3 bp, including one nonsynonymous replacement). The dendrogram based on the cyt b genes shown in Figure 2 suggests that P. ovale may be separated into three types. A similar branching pattern was obtained with the maximum likelihood method (data not shown). However, some sequence mistakes cannot be ruled out in GenBank-available sequences, such as that of Harding strain (for example, nt 202–221 are conserved in all reported Plasmodium spp. so far studied, except for P. ovale Harding strain and P. malariae Uganda-1). As a result, it seems more prudent to propose the separation of P. ovale into only two types. DiscussionBy analyzing the 3´ half of the P. ovale SSUrRNA genes, Li et al. (13) suggested that P. ovale might be separated into two types or subspecies, Nigerian I/CDC and LS. Later, the presence of LS-type or variant-type P. ovale was confirmed in Vietnam (4) and Africa (7); all variant-type isolates shared the same mutations at the block 9 in the SSUrRNA gene. Sequence analyses of the ookinete surface antigen gene, presented here and elsewhere (18), and of the cysteine protease gene all confirmed the occurrence of two different sequences in nuclear genes of parasites grouped as variant type and Nigerian I/CDC or classic type based on their SSUrRNA gene sequence. Whether the different sequences of SSUrRNA genes we describe for classic-type and variant-type P. ovale isolates correspond to A genes or include S or O genes is unclear (5,29). In P. falciparum (30) and P. vivax (5), extensive pairwise sequence diversity (>13% difference) has been reported between A and S or O genes. In both classic type and the variant isolates, however, SSUrRNA gene sequences were quite similar to each other (<4% difference), which suggests that they may all correspond to A genes. The occurrence of different A gene–like sequences may be a distinctive feature of P. ovale, indicating a possible field for future research. Because of the strict sequence conservation of the mitochondrial cyt b gene in natural isolates of the human malaria parasites P. falciparum and P. vivax, the divergence we found between sequences from variant- and classic-type parasites are putatively of major importance in defining two genetically distinct types of P. ovale. Analyzing the SSUrRNA gene of the Harding strain and determining whether this strain belongs to the variant-type or classic-type group or a third, poorly characterized group would be of interest. The prevalence and geographic distribution of P. ovale, the last human malaria parasite to be described, have elicited little interest until recently. We have previously shown that P. ovale is a widespread human pathogen in Southeast Asia (1,2); here we suggest that, in both Southeast Asia and Africa, at least two different types of P. ovale circulate in human hosts. This situation is reminiscent of that recently described for P. vivax, which may be divided into two types occurring respectively in the Old World and the New World (31). However, both variants of P. ovale (in contrast to those of P. vivax) occur in sympatry, which suggests that the genetic differentiation between them is not associated with geographic isolation. Moreover, the fact that human infections with variant-type P. ovale tend to be associated with higher levels of parasitemia, when compared with levels associated with classic-type parasites (2,4,11), may be the result of more dramatic biologic differences between these types, with possible clinical implications. Acknowledgments
References
|
||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||
|
||||||||||||
|
|
|
EID Home | Top of Page | Ahead-of-Print | Past Issues | Suggested Citation | EID Search | Contact Us | Accessibility | Privacy Policy Notice | CDC Home | CDC Search | Health Topics A-Z |
||
|
This page posted June
3, 2004 |
||
|
Emerging
Infectious Diseases Journal |
||