Preface
Introduction
1. The Clinical Trial Process
2. Clinical Trial Design and Interpretation of Results
3. Advancing Cancer Care Through Clinical Trials
4. Participant Protection in Clinical Trials
5. Barriers to Clinical Trial Participation
6. Conducting, Referring to, and Locating Clinical Trials
7. Case Study
Glossary
Bibliography
Preface
The public understands very little about clinical trials. Some
people are fearful of being "guinea pigs," even though participants
in clinical trials receive high-quality care. Other people are not
aware of clinical trials as an option, do not understand how they
work, or do not have access to them.
Likewise, health care professionals may be unaware of appropriate
clinical trials, may not want to refer people out of their practice,
may believe that standard therapy is best, or may think that getting
involved in clinical trials will add an undue administrative burden
to their work.
Today's standard cancer treatments were yesterday's clinical
trials. Successful clinical trials have:
Increased survival rates of participants with
testicular cancer, breast cancer, leukemia, and lymphoma
Decreased morbidity associated with the surgical
treatment of many cancers
Resulted in the development of new compounds and
techniques to reduce the side effects of cancer therapies
This guide is designed to familiarize health care professionals
and others with the ins and outs of clinical trials. It describes
how:
The clinical trial process works
Trials are designed to obtain particular information
Clinical trials advance standard cancer treatment
Trial participants are safeguarded
People might face obstacles to participating in clinical
trials
To find a local clinical trial
After reading the guide and reviewing the case study, the reader
will be better able to manage issues related to clinical trials. The
reader should be able to:
Discuss clinical trials as potential treatment or
preventive options
Answer people's questions and allay their fears about
clinical trials
Locate and refer people to accessible clinical
trials
Ultimately help advance the early detection, treatment,
and eventual prevention of cancer
Introduction
Approximately 555,550 people in the United States are expected to
die of cancer each year - an average of more than 1,500 people a day.
As the second leading cause of death after heart disease, cancer
accounts for one in four deaths each year. Moreover, about 1,284,900
new cancer cases are expected to be diagnosed in 2002. As
widespread as the threat of cancer is among all Americans, its impact
is felt disproportionately by racial and ethnic minorities, the
medically underserved, and people over age 65.
Scientific research continues to provide valuable insights into
the causes of cancer. But research is an incremental process, moving
forward in small, carefully planned steps. Advances typically begin
with basic research in the laboratory. After years of testing in
cells and tissues, promising leads are tested in animal models of
human cancers. Only after treatments or techniques prove successful
in animals can they be evaluated in people through clinical trials.
Well-designed, well-run clinical trials are the only way to determine
the true effectiveness of a promising new agent or intervention being investigated.
Clinical trials are designed to answer specific questions about
the effects of a therapy or technique designed to improve human
health. The trials are planned in advance, follow a rigorous
scientific process, and the findings are analyzed. The scientific
process has built-in safeguards for participants, who are selected
carefully from volunteers. Clinical trials are usually conducted in a
progressive series of steps, called phases. The process starts with
small trials testing the safety of an intervention and moves to
progressively larger trials. The larger trials compare the effectiveness of the new
intervention given to the investigational group to the currently accepted standard care given to the control group.
Clinical trials are mechanisms for developing better methods of
detecting, treating, and eventually preventing diseases like cancer.
The enormous strides made in treating childhood cancer, for example,
are the direct result of clinical trials. In the United States today,
more than 70 percent of children with cancer live at least 5 years
after diagnosis, as opposed to only 55 percent in the mid-1970s.
More than 60 percent of children with cancer participate in
clinical trials, yet only 3 percent of adults with cancer do. To
answer the most pressing questions about cancer - and to do so
quickly - many more adults must participate in clinical trials. To
encourage participation, the National Cancer Institute (NCI) and
other organizations provide information to ensure that health care
professionals and the people they treat understand clinical trials,
consider them as an option, and can easily locate them in their
communities. Clinical trials should not be considered only in terms
of caring for people who have cancer. They may also present
prevention and early detection options for people at high risk of
developing cancer.
For more basic information about clinical trials, see
"Facts and Figures about Cancer Clinical Trials 1."
1. The Clinical Trial Process
|
Learning Objectives
Identify the steps in the drug development process
Name the various types and phases of clinical trials
Describe special access programs
|
Clinical trials are a key part of the drug development and
approval process. The entire process takes place under the watchful
eye of the Food and Drug Administration (FDA). As a consumer
protection agency of the U.S. Department of Health and Human
Services, FDA is required by law to review all test results for new
drugs to ensure that they are safe and effective for specific uses.
"Safe" does not mean that the product is free of possible adverse
side effects; rather, it means that its potential benefits outweigh
any known risks. The FDA approval process is focused on drugs, but
similar processes exist for the approval of:
New devices (e.g., infusion pumps)
Agents (e.g., vitamins and medications)
Biologics (e.g., vaccines)
For purposes of illustration, the process outlined in this text
focuses on drug approval.
Before a new drug or biologic agent that shows promising results
in the lab can be tested in people, its sponsor must submit an
Investigational New Drug (IND) application to FDA. Once the
application is approved, the sponsor can begin testing the drug in
clinical trials with human participants. If these trials demonstrate
that the new drug is safe and superior to standard treatment, the
sponsor can file a New Drug Application (NDA) or a Biologics License
Application (BLA) to FDA. Only after FDA approves the drug can it be
marketed.
For an overview of the drug approval process from start to
finish, see
FDA's From Test Tube to Patient: New Drug Development in the United States 2. This book tells the story
of new drug development in the United States and highlights the
consumer protection role of FDA. For more information call 1-888-INFO-FDA
or visit the FDA's Web site 3.
|
Steps in the Drug Development Process
Early research and preclinical testing. During early
research and preclinical testing, drugs undergo basic
laboratory investigation and animal model testing for
efficacy and toxicity. This step takes about 4 years. Investigational New Drug application. The trial
sponsor files an Investigational New Drug (IND) application
with FDA. If FDA approves the application, clinical trials
begin. Phase 1 clinical trial. Phase 1 trials determine the
safety and appropriate dosage of the drug for humans. It
might take about 2 years before enough participants enroll
in the trial. If phase 1 trials are successful, researchers
design phase 2 trials. Phase 2 clinical trial. Phase 2 trials evaluate the
effectiveness of the drug and look for side effects. It
might take up to 2 years to enroll participants for these
trials. If phase 2 trials are successful, researchers design
phase 3 trials. Phase 3 clinical trial. Phase 3 trials evaluate the
effectiveness of the new treatment, compared to standard
treatment. It might take 3 to 4 years to enroll enough
participants for these trials. Researchers report trial
results in peer-reviewed scientific journals and at
professional meetings. New Drug Application. The trial sponsor files an NDA
or BLA with FDA. The sponsor submits this application to FDA
once it has adequate data to support a certain indication
for a drug (usually by finding that the drug is safe and
superior to standard treatment in a definitive phase 3
trial). FDA approval. FDA approves the claim that is
being made about the drug, which takes up to about 1
years. After approval, it can be marketed to the public.
FDA approval allows the drug to be "labeled" for a
specific use. This label includes information on the
drug's dosage, indications, safety, and side effects.
|
Cancer clinical trials focus on developing new strategies for the
prevention, detection, treatment, and overall improvement of the care
and quality of life of people with cancer or people at high risk for
developing cancer. In cancer research, a clinical trial is designed
to show how a particular anticancer strategy affects the people who
receive it.
Clinical trials differ by type and phase, but they all involve
rigorous scientific testing. Each type of clinical trial attempts to
answer different research questions:
Prevention trials: What kinds of interventions -
such as lifestyle modifications, dietary supplements, or drugs -
can prevent cancer from occurring?
Screening and early detection trials: What tests can
find cancer as early as possible in healthy people?
Diagnostic trials: How can new tests or procedures
identify a suspected cancer earlier or more accurately?
Genetics trials: Can gene-transfer therapy be used to
treat cancer?
Treatment trials: What new interventions (e.g., drugs,
biologics, surgical procedures, radiation) can help people who
have cancer?
Quality-of-life and supportive care trials: What kinds
of interventions can improve the comfort and quality of life of
people who have cancer?
Clinical trials occur in four phases, each of which is designed to
answer different research questions:
Phase 1: How does the treatment affect the human
body? How should the treatment be given? What dosage is safe?
Phase 2: Does the treatment do what it is supposed to do
for a particular cancer? How does the treatment affect the human
body?
Phase 3: Is the new treatment (or new use of a
treatment) better than current practice?
Phase 4: What are the effects of an approved
treatment?
The phases of clinical trials are explained in the context of drug
treatment trials on the pages that follow. But the same concepts
apply to most types of clinical trials, which are described after
treatment trials.
|
filler
|
Phase
1
|
Phase
2
|
Phase
3
|
Phase
4
|
|
Number of participants
|
15-30 people
|
Less than 100 people
|
Generally, from 100 to thousands of people
|
Several hundred to several thousand people
|
|
Purpose
|
*To find a safe dosage
*To decide how the agent should be given
*To observe how the agent affects the human body
|
*To determine if the agent or intervention has an
effect on a particular cancer
*To see how the agent or intervention affects the
human body
|
*To compare the new agent or intervention (or new
use of a treatment) with the current standard
|
*To further evaluate the long-term safety and
effectiveness of a new treatment
|
Treatment Trials
Treatment trials are designed to test the safety and effectiveness
of new drugs, biological agents, techniques, or other interventions
in people who have been diagnosed with cancer. These trials evaluate
the potential clinical usefulness of a therapy or compare an
investigational treatment against standard treatment, if there is
one.
Phase 1
Phase 1 trials are the first step in transforming laboratory data
into clinical care. While the primary goal of a phase 1 trial is to
determine the toxic effects, pharmacological behavior, and
recommended dosage of a therapy or technique for future trials, these
trials are conducted with therapeutic intent.
In a phase 1 trial, the study participants (usually less than 30
people) are divided into cohorts of three to six participants. Each
cohort of participants is treated with an increased dose of the new
therapy or technique. Results in early participants greatly influence
the dose subsequent participants receive. Initial dosage is based on
preclinical testing and is usually quite conservative. If no serious
side effects are seen in the initial group after a period of time,
usually 3 to 4 weeks, the next group of participants receives a
higher dose. This pattern is repeated until a certain percentage of
participants experience dose-limiting toxicity - that is, side
effects strong enough that the next group of participants should not
get a higher dose. The highest dose with acceptable toxicity is
determined to be appropriate for further testing.
Phase 1 trials are not limited to "first in human" studies.
Subsequent phase 1 trials often evaluate new schedules or
combinations of established drugs or radiation. Later phase 1 trials
may also be conducted to evaluate toxicity, response, and
pharmacokinetics in populations that might not have been included in
prior trials, such as children or the elderly. Some phase 1 trials
are pilot trials for larger trials designed to determine the
interaction of a drug with another treatment or substance.
Who Participates
Almost all phase 1 trials of new anticancer drugs involve
participants with a cancer that lacks or does not respond to
standard treatment. People with many types of cancer can
participate in the same phase 1 trial. Participants are generally
required to have organ function capable of metabolizing and
excreting the drug and a 1- to 2-month life expectancy, in order
to evaluate the drug's effects and the body's response to it.
Possible Benefits
Possible Risks
Phase 2
Phase 2 trials are designed to evaluate the effectiveness of the
drug in a somewhat larger group of participants (usually under 100),
using the dosage determined to be safe in phase 1 trials. On the
basis of their findings in phase 1 trials, researchers often focus
phase 2 trials on cancers for which no effective treatment exists
and/or that are most likely to show a response to therapy. In
choosing which type of cancer to study, researchers may also take
into account effective alternatives and choose a cancer that has
none. Some anticancer compounds being developed target molecular
pathways in specific cancers, a development that may affect the
cancers chosen for phase 2 trials.
In most phase 2 trials, all participants receive the same dose of
the drug (or undergo the same intervention). The new treatment is
assessed for effectiveness, and additional safety information is
noted. Even if the new treatment seems effective, it usually requires
further testing before entering widespread use. Because the treatment
has not been compared to any other therapy or technique, its relative
value is unclear, and it is impossible to rule out other factors that
may have influenced its effectiveness. In addition, phase 2 trials
are often too short to determine long-term benefits; larger and
longer phase 3 trials are more suited to this purpose.
Some phase 2 trials compare different schedules of administering
the same drug. At the end of such trials, the most promising regimen
is chosen to move into phase 3 trials. Participants in this type of
phase 2 trial are assigned at random to either the investigational
group, which is given the new treatment, or the control group, which
receives the standard treatment. Neither the participants nor their
doctors choose which group individual participants will be in.
Who Participates
Generally, people who take part in phase 2 trials have
not found the current standard of care effective or have cancers
for which there is no standard treatment. Participants are
generally required to have adequate organ function, a 3-month life
expectancy, and a limited number of prior treatments.
Possible Benefits
Possible Risks
Phase 3
Phase 3 trials are large trials (usually involving more than 100
participants) designed to determine whether a new therapy or
technique is more effective or less debilitating than a standard
treatment. These trials are conducted at multiple institutions around
the country, including community settings. Because the results of
phase 3 trials guide health care professionals and people with cancer
in making treatment decisions, their results should apply to aspects
such as survival time and quality of life.
Like phase 2 trials, phase 3 trials usually focus on specific
types of cancer. Participants enrolling in a phase 3 trial are
assigned at random to an investigational group, which is given the
new treatment, or a control group, which receives the current
standard treatment. Some trials can also include more than two study
groups, depending on the research questions being asked.
Who Participates
Many people with cancer get their first treatment in a
phase 3 trial. Eligibility requirements vary with the disease
stage or other factors being studied. Phase 3 trials typically
involve large numbers of participants in order to determine true
effectiveness.
Possible Benefits
Regardless of the group a participant is assigned
to, he or she will receive at a minimum the best widely accepted
standard treatment.
If a participant is taking the new treatment and it is
shown to work, he or she may be among the first to benefit.
Possible Risks
New treatments under study are not always better
than, or even as good as, standard treatment.
New treatments may have side effects that are worse than
those of standard treatment.
Despite phase 1 and 2 testing, unexpected side effects
may occur.
If the new treatment has benefits, it still may not work
for every participant (just as standard treatments do not help
everyone).
Participants receiving the standard treatment may not
benefit as much as those receiving the new one.
|
Finding Out About Standard Cancer Care
Standard cancer care is the accepted and widely used treatment for a certain type of cancer. It is based on the results of past research. The National Cancer Institute's Web site
www.cancer.gov
contains a database of the latest information about cancer
and clinical trials. Specialists review current literature
from more than 70 medical journals, evaluate its relevance,
and synthesize it into clear summaries for the public and
health professionals. Many of the summaries are also
available in Spanish.
|
Phase 4
Phase 4 trials are used to further evaluate the long-term safety
and effectiveness of a treatment. Less common than phase 1, 2, and 3
trials, phase 4 trials usually take place after the new treatment has
been approved for standard use.
Other Types of Trials
Adjuvant and Neoadjuvant Treatment Trials
Adjuvant trials are additional therapy after standard treatment.
They are designed to prevent the recurrence of cancer in people who
no longer show clinical evidence of disease. Adjuvant trials attempt
to treat the subclinical or microscopic disease thought to be
responsible for cancer recurrence and therefore improve disease-free
and overall survival. The combination of standard and adjuvant
treatments is initially tested in a small feasibility or pilot study
similar to a single-agent phase 2 trial. This is followed by a
randomized phase 3 trial if the combination proves safe and
effective.
Neoadjuvant trials are additional therapy before standard
treatment. These trials evaluate treatments designed to reduce tumor
size to a point where it can be definitively treated by therapies
that are considered the best standard treatment. For example,
clinical trials have shown that chemotherapy can reduce an inoperable
breast cancer to a size that can be removed surgically.
Both adjuvant and neoadjuvant trials are phased like other
treatment protocols, with the phase dependent on the major objective
of the trial.
Who Participates
People who have no clinical evidence of disease after
primary treatment, but who are at high risk of recurrence,
participate in adjuvant trials. People whose cancer, once reduced,
could be effectively treated by therapies considered the best
standard treatment participate in neoadjuvant trials.
Prevention Trials
Cancer prevention trials are designed for people at risk of
developing cancer. The trials evaluate the safety and effectiveness
of various risk reduction strategies. The two types of prevention
trials answer the following questions:
Action trials: Can a person's actions - such as
exercising more or quitting smoking - prevent cancer?
Agent trials: Can taking certain medicines, vitamins,
minerals, or food supplements lower the risk of certain types of
cancer?
(Agent trials are also known as chemoprevention trials.)
Chemoprevention trials compare a promising new prevention agent or
technique with a standard agent or technique, or placebo. The
investigational group takes the agent being studied; the control
group takes either the standard agent that is being compared to the
study agent or - because there may be no standard agent - a
look-alike agent that contains no active ingredient, called a
placebo.
Who Participates
Prevention trials seek participants from various age
groups and socioeconomic backgrounds or people who have
combinations of cancer risk factors. Participants in prevention
trials are otherwise healthy individuals who are at risk for
cancer.
Possible Benefits
Possible Risks
New cancer prevention interventions may have
unknown side effects or risks.
The drug intervention may have worse side effects or be
less effective than standard preventive measures.
Even if a new drug or intervention is effective, it may
not work for every participant.
Screening Trials
Screening trials assess the effectiveness of new means of
detecting the earliest stages of cancer. In addition, these trials
examine whether early treatment improves overall survival or
disease-free survival. Screening tools include imaging tests and
laboratory tests.
Who Participates
Participants are healthy and may be chosen to represent
particular age groups or socioeconomic backgrounds. Screening
trials also seek participants with certain cancer risk factors,
such as belonging to a family that has a genetic predisposition to
cancer.
Possible Benefits
For many types of cancer, detecting the disease at
an early stage can result in earlier treatment and an improved
outcome.
Screening trials often encourage participants to
continue screening on a regular basis, which can lead to improved
health overall.
Screening trials for people with a genetic
predisposition to cancer can alert other family members to begin
regular cancer screening, aid in early detection, and help in the
diagnosis and treatment of potential cancers.
Possible Risks
Some of the imaging procedures used in screening
may be uncomfortable or require participants to be in confined
spaces for some period of time.
If an imaging technique is being studied, participants
may be exposed to x-rays or radioactive substances.
Tests can be time consuming.
Diagnostic Trials
Diagnostic trials develop better tools for physicians to use in
classifying types and phases of cancer, and in managing the care of
people with cancer. Some trials compare the ability of two diagnostic
techniques to provide information about a suspected cancer. Genetic
tests are being evaluated as diagnostic tools to classify cancers
further, thus helping physicians direct cancer therapy and improve
treatments for people with specific genetic mutations. Diagnostic
trials may also evaluate techniques designed to measure and monitor
cancer response more accurately or less invasively, such as using a
new imaging tool that eliminates the need for surgery.
Who Participates
Participants include people with cancer or symptoms
suggesting cancer.
Possible Benefits
The diagnostic test under investigation may be
better and less invasive than current tests.
A new diagnostic tool may help detect cancer recurrence,
which could lead to improved outcomes.
Possible Risks
Genetics Trials
Actual genetic intervention (such as gene-transfer) trials are few
in number, however trials are underway where actual cellular
manipulation at the gene level occurs. Most genetics research
involves looking at tissue or blood samples from large populations of
people in order to determine how genetic make-up can influence
detection, diagnosis, prognosis, and treatment. This genetic
epidemiologic research does not involve any actual intervention.
Rather, it is designed to broaden understanding of the causes of
cancer. Genetics research is also being used to develop targeted
treatments based on the genetics of a tumor.
Genetics research is a critical component of cancer research
because it helps scientists understand the causes of cancer and can
lead to developing clinical trials for the prevention, detection, and
treatment of cancer.
Quality-of-Life and Supportive Care Trials
Quality-of-life and supportive care trials test interventions
designed to improve quality of life for people with cancer and their
families. They seek better therapies or psychosocial interventions
for people experiencing nutrition problems, infection, pain, nausea
and vomiting, sleep disorders, depression, and other effects of
cancer or its treatment. Some supportive care trials target families
and caregivers to help them cope. The effectiveness of supportive
care trials may be measured either:
Who Participates
Possible Benefits
Possible Risks
Investigational drugs may be made available for use outside of a
clinical trial. Working with NCI and other sponsors, FDA has
established special conditions under which a person with cancer can
receive unapproved cancer drugs that have shown clinical benefit.
Group C
In the 1970s, NCI researchers became concerned about the time it
took to bring to market investigational drugs found to have antitumor
activity. Working with FDA, NCI established the "Group C"
classification to allow access to drugs with reproducible activity.
Group C agents are investigational drugs provided by the National Cancer Institute to properly trained physicians for the treatment of individual patients who meet specific eligibility criteria within this category and are treated according to a protocol.
Each Group C drug protocol specifies eligibility, reporting
methodology, and drug use. Group C designation speeds new drugs to
people who need them most. The process allows NCI to gather important
information on the safety as well as activity of the drugs in the
settings where they will be most used after FDA approval. Drugs are
placed in the Group C category by agreement between FDA and NCI.
Group C drugs are always provided free of charge, and the Centers for
Medicare and Medicaid Services (formerly the Health Care Financing
Administration) provides coverage for its beneficiaries for care
associated with Group C therapy.
Treatment IND
In 1987, FDA began authorizing the use of new drugs still in the
development process to treat certain seriously ill people. In these
cases, the process is referred to as a treatment Investigational New
Drug (IND) application. Clinical trials of the new drug must already
be underway and have demonstrated positive results.
FDA sets guidelines about:
What serious and life-threatening illnesses
constitute
How much must be known about a drug's side effects and
benefits
Where physicians can obtain the drug for treatment
For many seriously ill people, the possible benefits outweigh the
risks associated with taking an unapproved drug.
Less common ways that people can receive investigational drugs are
through expanded access protocols or mechanisms known as special or
compassionate exception.
Expanded Access Protocols
Expanded access protocols are available for a limited number of
well-studied investigational drugs awaiting final FDA approval.
Expanded access allows a wider group of people to be treated with a
drug. The purpose is to make investigational drugs that have
significant activity against specific cancers available before the
FDA approval process has been completed.
The IND sponsor must apply to FDA to make the drug available
through an expanded access protocol. There must be enough evidence
from completed trials to show that the drug may be effective to treat
a specific type of cancer and that it does not have unreasonable
risks. FDA generally approves expanded access only if no other
satisfactory treatments are available for the disease.
Special or Compassionate Exception
People who do not meet the eligibility criteria for a clinical
trial of an investigational drug may be eligible to receive the drug.
The person's doctor contacts the trial sponsor and provides the
person's medical information and treatment history; requests are
evaluated on a case-by-case basis. FDA must approve each request to
provide the drug outside a clinical trial. There should be reasonable
expectation that the drug will prolong survival or improve quality of
life.
Considerations when determining whether a person may receive an
investigational drug as a special exception include:
Is the person ineligible for a clinical trial?
Have standard therapies been exhausted?
Is there objective evidence that the investigational
agent is effective for the person's type of disease?
Can the drug potentially benefit the person?
What is the risk to the person?
In some cases, even people who qualify might not be able to obtain
the drug if it is in limited quantity and high demand.
Refer to the case study 4 for a review and summary of content covered in this workbook.
2. Clinical Trial Design and Interpretation of Results
|
Learning Objectives
Define key members of the research team
Review key components of a clinical trial
Describe the purpose of the randomization,
stratification, and blinding in clinical trial protocols
Name common statistical methods used to interpret
clinical trial results
|
Clinical trials follow strict scientific guidelines that dictate how
a study is designed and who participates in it. The reasons for these
guidelines may not be immediately clear to a person urgently seeking
treatment, but they protect people and provide scientifically sound
results that can lead to truly effective therapies and
techniques.
Designing and implementing a clinical trial requires the many
talents of a multidisciplinary research team. Each team may be set up
differently, depending on an institution's policy and resources.
Typical team members and their responsibilities include:
Principal investigator - oversees all aspects of a
clinical trial, specifically, concept development, protocol
writing, protocol submission for institutional review board (IRB)
approval, participant recruitment, informed consent, and data
collection, analysis, interpretation, and presentation.
Research nurse - coordinates the clinical trial and
educates staff, participants, and referring health care providers.
This nurse acts as an information conduit from the clinical
setting to the principal investigator and vice versa, and assists
the principal investigator with toxicity and response monitoring,
quality assurance, audits, and data management and analysis.
Data manager - handles the management of clinical trial
data, including electronic data entry. Collaborates with the
principal investigator and research nurse to identify what
participant data will be tracked. The data manager also provides
data to monitoring agencies and prepares summaries for interim and
final data analysis.
Staff physicians and nurses - administer treatment to
participants as specified in the protocol; assess and record
toxicity, drug tolerance, and adverse events; collaborate with the
principal investigator and research nurse in observing and
reporting clinical trends; and provide clinical management and
participant education.

Protocol
Every trial has a written, detailed action plan, called a
protocol. The protocol provides the background, specifies the
objectives, and describes the design and organization of the trial.
Every site participating in the trial uses the same protocol,
ensuring consistency of procedures and enhancing communication. This
uniformity ensures that results from all sites can be combined and
compared.
The clinical trial protocol answers the following questions:
What is the scientific rationale or basis for
conducting the trial?
What are the objectives?
How many participants will be in the trial?
Who is eligible to participate? (This is determined on
the basis of factors such as age and disease status.)
What is the intervention, and what is its duration or
schedule?
What side effects might there be?
What medical tests or followup visits will participants
have? How often?
What information will be gathered about
participants?
What are the endpoints of the trial?
The following FDA-required protocol elements help investigators
answer the questions above and assist participants and health care
professionals in understanding the goals of a clinical trial:
General information
Background information (with relevant references from
the scientific literature)
Trial objectives and purpose
Trial design
Participant selection and withdrawal
Participant treatment
Efficacy assessment
Safety assessment
Statistics
Direct access to source data and documents
Quality control and quality assurance
Ethics
Data handling and record keeping
Financing and insurance
Publication policy
Supplements
Eligibility Criteria
Participant eligibility criteria can range from general (age, sex,
type of cancer) to specific (prior treatment, tumor characteristics,
blood cell counts, organ function). Eligibility criteria may also
vary with trial phase. In phase 1 and 2 trials, the criteria often
focus on making sure that people who might be harmed because of
abnormal organ function or other factors are not put at risk. Phase 2
and 3 trials often add criteria regarding disease type and stage, and
number of prior treatments.
Eligibility criteria might be very detailed if researchers think
that a drug will work best on a specific type of cancer or
population. Trials with narrow eligibility criteria might be
complicated to conduct and might produce less widely applicable
results.
Researchers therefore attempt to include as many types of people
as possible in a clinical trial without making the study population
too diverse to tell whether the treatment might be as effective on a
more narrowly defined population. The more diverse the trial's
population, the more useful the results could be to the general
population, particularly in phase 3 trials. Results of phase 3 trials
should be as generally applicable as possible in order to benefit the
maximum number of people.
The trend today is toward broadening eligibility criteria for
phase 3 clinical trials. Less restrictive criteria may enable more
researchers and people with cancer to participate in these trials.
With more participants, the disadvantages of having a more diverse
population will be outweighed by the results applying more generally
to the population.
Endpoints
An endpoint is a measurable outcome that indicates an
intervention's effectiveness. Endpoints differ depending on the phase
and type of trial. For instance, a treatment trial endpoint could be
tumor response or participant survival. Quality-of-life or supportive
care trial endpoints could include participants' welfare and control
of symptoms.
Examples of endpoints include:
Tumor response rate - the proportion of trial
participants whose tumor was reduced in size by a specific amount,
usually described as a percentage. If 7 of 10 patients responded,
the response rate is 70 percent.
Disease-free survival - the amount of time a participant
survives without cancer occurring or recurring, usually measured
in months.
Overall survival - the amount of time a participant
lives, typically measured from the beginning of the clinical trial
until the time of death.
Tumor response rate is a typical endpoint in a phase 2 treatment
trial. However, even if a treatment reduces the size of a
participant's tumor and lengthens the period of disease-free
survival, it may not lengthen overall survival. In such a case, side
effects and failure to extend overall survival might outweigh the
benefit of longer disease-free survival. Alternatively, the
participant's improved quality of life during the tumor-free interval
might outweigh other factors.
Because tumor response rates are often temporary and may not
translate into long-term survival benefits for the participant,
response rate is a reasonable measure of a treatment's effectiveness
in a phase 2 trial, whereas participant survival and quality of life
are better endpoints in a phase 3 trial.
Randomization
In phase 3 trials (and some phase 2 trials) participants are
assigned to either the investigational or control group by chance,
via a computer program or table of random numbers. This process,
called randomization, gives each person the same chance of being
assigned to either group. Randomization ensures that unknown factors
do not influence the trial results.

|
Randomization is a method used to prevent bias in research. A
computer or a table of random numbers generates treatment
assignments, and participants have an equal chance to be assigned to
one of two or more groups (e.g., the control group or the
investigational group).
|
If physicians or participants themselves chose the group,
assignments might be biased. Physicians, for instance, might
unconsciously assign participants with a more hopeful prognosis to
the experimental group, thus making the new therapy seem more
effective than it really is. Conversely, participants with a less
hopeful prognosis might pick the experimental treatment, leading it
to look less effective than it really is.
Randomization tends to produce comparable groups in terms of
factors affecting prognosis and other participant characteristics. In
this way, randomization guarantees the validity of the conclusion
concerning the effectiveness of the treatment.
Stratification
Stratification is used in randomized trials when factors that can
influence the intervention's success are known. For instance,
participants whose cancer has spread from the original tumor site can
be separated, or stratified, from those whose cancer has not spread.
Assignment of interventions within the two groups is then randomized.
Stratification enables researchers to look at factors in both
groups.
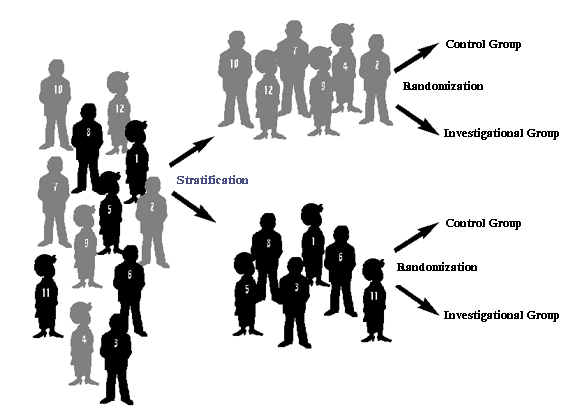
|
|
Stratification is a process used in randomized trials when
factors that can influence the intervention's success are known.
Assignment of interventions within the two groups is then randomized.
Stratification enables researchers to look in separate subgroups to
see whether differences exist.
|
Blinding
Trials set so that participants do not know which intervention
they are receiving are known as single-blinded trials. Those in which
neither researchers nor participants know who is in the
investigational or control group are called double-blinded trials.
Double-blinded trials ensure that people assessing the outcome will
not be influenced by knowing which intervention a participant is
receiving and also that ancillary followup treatment will be the
same.
Data Collection and Management Tools
Most research teams use standardized and newly created tools to
collect, process, analyze, and audit data. Tools vary in format from
visual analog scales to open-ended questionnaires. Examples of tools
for participants to use to self-report data include diaries,
calendars, logs, and surveys.
The case report form is the basic tool of data abstraction. Many
reports use a Web-based format, others are paper-based. NCI is
constructing an informatics system that will reduce the extensive
paperwork often associated with clinical trials. For example, the
Common Toxicity Criteria (CTC), a Web-based, interactive application,
uses standardized language to identify and grade adverse events in
cancer clinical trials. Forms are also available for rapid reporting
of adverse events, electronically or by telephone, to alert
researchers to potential safety issues. The Adverse Event Expedited
Reporting System (AdEERS) is a Web-based program that enables
researchers using NCI-sponsored investigational agents to expedite
the reporting of serious and/or unexpected adverse events directly to
NCI and FDA.
Researchers use statistical methods to determine whether an effect
observed in a clinical trial is real (statistically significant) or
caused by chance (not statistically significant). Although the
examples included here use terminology and illustrations from
treatment trials, these statistical techniques apply to all types of
clinical trials.
Key Terms
Familiarity with the following terms is useful in understanding
how researchers use statistics to interpret clinical trial
results:
p-values reflect the likelihood that the results
of a clinical trial are because of chance rather than due to a
real difference between the tested treatments. The smaller the
value of p, the greater the likelihood that the results are not
because of chance. A p-value of 0.05 (that is, 1 in 20) or smaller
is widely accepted as an indication that the results are
statistically significant.
Confidence intervals reflect a range of values of the
true value that would be obtained if everyone with a particular
cancer were treated with the treatment under study. The wider the
interval, the more variable the result and the less likely it is
to be close to the true value. Confidence intervals are typically
thought of as the approximate bounds or limits of the true value.
Researchers frequently use either a 95 or a 99 percent confidence
interval.
Sample size is the number of people participating in a
trial.
Statistical power refers to the chance of finding a
statistically significant result when there is one. Ideally,
statistical power should be 0.80 or 0.90 - reflecting an 80 to 90
percent chance of detecting that the true difference in treatment
effectiveness is the smallest size considered medically important
to detect.
Relative risk is the likelihood that cancer will occur
within a specific timeframe in one group versus another.
Statistical Significance
The result of a clinical trial can be statistically significant
(not due to chance) without being clinically significant (medically
important). Suppose, for instance, that a group receiving an
experimental treatment has a 2 percent higher survival rate than the
group receiving the standard treatment. This difference could be
statistically significant, but if participants who survive longer
experience serious side effects, it may not be medically important.
In this case, the side effects might be worth tolerating only if the
experimental treatment group has a 10 percent higher survival rate.
Good trial planning and interpretation take into consideration both
medical importance and statistical significance.
The results of a trial are usually considered statistically
significant when data comparison results in a p-value of 0.05 or
smaller. If the p-value is 0.01 or even 0.001, the results are
considered even more significant because there is less likelihood
that the results are due to chance.
Confidence intervals are often useful data for researchers because
they enable researchers to generalize the results of the trial to the
population.
For example, in a treatment trial with an investigational and a
control group, the mean (average) values of the endpoints (e.g.,
survival for 5 years after treatment) are calculated separately for
each group. Then the standard error - how far the values extend on
either side of the mean - is calculated for each group. The less
overlap between the confidence interval for the standard treatment
group and the experimental treatment group, the more likely the
difference between the groups is statistically significant. Research
reports typically include confidence intervals, for example:
The rate of 5-year survival for group A was 73% (95%
confidence interval, 65.7% to 80.3%). The rate of 5-year survival
for group B was 58% (95% confidence interval, 49.8% to 66.2%). p =
0.004.
In this case, the confidence intervals come close to each other -
65.7 percent and 66.2 percent - but do not overlap. The p-value is
definitely statistically significant.
Confidence intervals can give an indication of whether the results
of small-sized trials that are not statistically significant are
nevertheless medically significant. They can be particularly
important tools when the trial size is limited because the type of
cancer is rare.
Trial Size
The number of participants in a clinical trial greatly influences
its statistical significance. With too few participants, a trial does
not generate enough information to draw a conclusion, and important
results may be missed. On the other hand, by testing more people than
needed to obtain statistically significant results, a trial takes
longer to produce results and may give ineffective or unsafe therapy
to more people than necessary.
When planning a clinical trial, researchers first decide how large
a difference between treatment groups is medically important. Next,
they calculate sample size, or how many people should be enrolled in
the trial. The sample should include enough participants to get a
statistically significant result (a p-value of 0.05 or smaller).
Sample size also influences the statistical power of the research
and is calculated before the trial begins. As sample size increases,
statistical power increases. Ideally, power should be 0.80 or 0.90.
Calculating statistical power helps a researcher decide how many
people to enroll in a trial.
Relative Risk
Relative risk usually describes the risk of getting cancer based
on lifestyle, environmental exposure to cancer-causing agents, or
family history of disease. However, when used in cancer clinical
trial reports, relative risk usually indicates the likelihood that cancer will
occur within a specific timeframe in one group versus another.
Intention to Treat
Phase 3 trials are often analyzed on an intention-to-treat basis -
that is, all participants who were initially admitted into the trial
and randomized are included in the primary analysis.
Intention-to-treat analysis therefore includes people who:
Did not follow instructions
Can no longer be located or contacted
Withdrew from the trial
Did not receive treatment
Including data from the groups above may weaken the results of a
trial, but excluding the data would bias the trial. For instance, if
half of the people in a treatment group withdrew because they thought
the drug they were taking was ineffective and had severe side
effects, and if the other half of the group had a 50 percent response
rate, then excluding the data from the participants who withdrew
makes the drug appear to be 50 percent effective. The actual response
rate is 25 percent. Intention-to-treat analysis typically excludes
participants who were ineligible to be included in the trial but were
randomized.
Refer to the case study 4 for a review and summary of content covered in this workbook.
3. Advancing Cancer Care Through Clinical Trials
Learning Objectives
Describe the FDA drug approval process
Describe how clinical trial results are released
Describe clinical trials that have led to advances
in cancer prevention, detection, and treatment
Discuss the importance of professional referral
and patient participation in the research process
|
Once phase 1 and 2 treatment trials are completed, the data are
analyzed and, if the treatment shows promise, it moves into phase 3
trials. As soon as the treatment sponsor thinks that phase 3 data
show it is safe and superior to standard treatment, the sponsor may
submit a New Drug Application (NDA) or a Biologics License
Application (BLA), to FDA for approval. At this stage, FDA approves
only the claim being made about the drug or intervention, not the
drug or intervention itself
A New Drug Application includes:
The exact chemical or biological makeup of the
therapy and the mechanisms by which it is thought to be effective
Results of animal studies
Results of clinical trials
How the drug or therapy is manufactured, processed, and
packaged
Quality control standards
Information about drug or intervention samples of the
product in the form(s) in which it is to be administered
FDA assesses applications in order of importance, giving first
priority to interventions with the greatest potential benefits. All
drugs that offer significant medical advances are considered priority
drugs in the approval process.
Independent advisory committees of professionals from outside the
agency give expert advice and guidance in making decisions about drug
approval. By law, these committees include both a patient
representative and a consumer representative. One such committee is
the Oncologic Drugs Advisory Committee, which meets regularly to
consider most cancer-related treatments and preventive drugs. The
committee assesses the safety, effectiveness, and appropriate use of
products considered for approval.
As FDA looks at the sponsor's data and its own review results, it
applies two questions to each application:
Do the results of well-controlled clinical trials provide
substantial evidence of effectiveness? Do the results show that the product is safe under the proposed
conditions of use? (In this context, "safe" means that
potentialbenefits outweigh any known risks.)
Once FDA has approved a new drug, the drug is "labeled" for a
specific use. This label includes information on eligibility, dose,
safety, and adverse effects. The agency's responsibility for new
treatments does not stop with final approval. FDA also:
Implements and tracks programs to make sure
manfacturers comply with standards and practice regulations
Monitors new drug advertising to make sure it is
truthful and complete
Handles feedback from health professionals and consumers
about effectiveness, adverse reactions, and potential problems in
labeling and dosage
The results of a clinical trial are usually reported first in
peer-reviewed scientific journals. If the results appear to have
significant medical importance, researchers make a public
announcement when the formal report is submitted for publication,
ensuring that people benefit from the new treatment as soon as
possible. Particularly important results are featured by the media
and widely discussed at scientific meetings and within advocacy
groups.
Clinical trial results are not available to the public as the
trial progresses because:
Knowing interim results could influence medical
personnel and participants in the trial, biasing the results
Statistical analysis might be less meaningful,
compromising the accuracy of the findings
In the absence of very clear evidence that a trial should be
stopped early for medical reasons, trials are completed before
reporting results. Interim results are unavailable to the public, and
often to the research teams. Independent data and safety monitoring
boards track phase 3 trial data. These boards alert researchers about
any safety or effectiveness issues that arise during the trial. Data
and safety monitoring plans are also in place for many phase 1 and 2
trials.
Research progresses in small steps, and sometimes publishing the
results of a trial is not as important as taking what was learned
from the trial and building on it in a new trial. To find the results
of a clinical trial, search a medical literature database, like
Medline or PubMed, available online through the National Library of
Medicine (www.nlm.nih.gov) and at medical institutions' libraries.
The "closed protocol" file in NCI's PDQ® (Physician Data Query)
may contain related studies (see section 6 for more information on
PDQ).
It often takes more than a year for a scientific paper to be
written, submitted, reviewed, edited, and published. If an initial
literature search turns up nothing, try again after some time has
passed.
Once an intervention is proven safe and effective, it may become
the new standard of care. Thus, current cancer care is based on the
results of past clinical trials. Recent clinical trials have resulted
in the following treatment benefits for people with chronic
myelogenous leukemia, cervical cancer, breast cancer, and
melanoma.
Chronic Myelogenous Leukemia - A New Treatment Option
In 2001, FDA approved GleevecTM, offering a new treatment
option for many people with chronic myelogenous leukemia (CML).
Until then, bone marrow transplantation in the initial chronic
phase of the disease was the only known effective therapy for CML.
However, this is not an option for many people and the procedure
can cause serious side effects or death. Another option, treatment
with the drug interferon alfa, may produce remission (a decrease
in or disappearance of signs and symptoms of cancer) for many
people. But if the drug is ineffective or people stop responding
to it, their prognosis is generally bleak.
In three short-duration, early-phase clinical trials with
Gleevec, researchers found that people with CML either had higher
remission rates than expected or they had few side effects.
Gleevec was designed to target an abnormal version of a cellular
protein present in nearly all people with CML. The abnormal
protein is much more active than the normal version and probably
causes the disease. By blocking the abnormal protein, called
BCR-ABL, Gleevec kills the leukemia cells.
Gleevec represents a new class of cancer drugs, which target
abnormal proteins that are fundamental to the cancer itself.
Cervical Cancer - Improved Survival Rates
For many years, the standard therapy for invasive
cervical cancer was surgery or radiation alone. Five large
clinical trials showed that women with invasive cervical cancer
have improved survival rates when they receive a
cisplatin-containing chemotherapy regimen plus radiation therapy.
Breast Cancer
Less Extensive Surgery, Same Survival Time
For many years, the standard therapy for all breast cancers was
a modified radical mastectomy with radiation or chemotherapy.
Clinical trials showed that for women with early-stage disease,
long-term survival after lumpectomy with axillary lymph node
dissection plus radiation therapy is similar to survival after
modified radical mastectomy.
Reduced Risk for Women at High Risk
Traditionally, women seeking to reduce their risk of breast
cancer had no clear option. A large phase 3 clinical trial
assessed risk reduction in women taking the drug tamoxifen. The
trial found that high-risk women who took the drug for up to 5
years (an average of 4 years) had 49 percent fewer diagnoses of
invasive breast cancer than those taking a placebo.
Melanoma - Improved Survival
According to the findings of a large, randomized clinical
trial, compared to low-dose interferon or no therapy, high-dose
interferon alfa-2b (Intron-A) significantly prolongs disease-free
survival for people at high risk for melanoma recurrence.
Biological Therapy
Biological therapy (sometimes called immunotherapy, biotherapy, or
biological response modifier therapy) uses the body's immune system,
either directly or indirectly, to fight cancer or to lessen the side
effects that some cancer treatments might cause.
The immune system is a complex network of cells and organs that
work together to defend the body against attacks by "foreign," or
"nonself," invaders. This network is one of the body's main defenses
against disease. It works against disease, including cancer, in a
variety of ways. For example, the immune system may recognize the
difference between healthy cells and cancer cells in the body, and
work to eliminate those that become cancerous. Biological therapies
are designed to repair, stimulate, or enhance the immune system's
responses. Many clinical trials are testing the use of biological
therapies, such as monoclonal antibodies and vaccines, to treat
cancer.
Monoclonal Antibodies
Monoclonal antibodies (MOABs) are a form of biological therapy now
being studied in the laboratory and in clinical trials.
MOABs are designed to fill a critical gap in the body's immune
system. Although the human body naturally produces antibodies to
identify and fight off viral and bacterial infections, the immune
system may not always recognize cancer cells as harmful. This is
because some cancer cells do not possess an antigen on their cell
membrane that is capable of eliciting an immune response. Therefore,
cancer is able to grow and spread unchecked. MOABs are being
developed to supplement the body's immune system by recognizing and
attacking specific proteins that cancer cells express. These specific
antibodies may be active on their own, or they may be linked to a
drug to allow specific delivery of the drug to the cancer cell.
Basic immunologic research identified a molecule specific to the
surface of B-lymphocytes that also is highly expressed on the surface
of most lymphomas. An antibody directed against this molecule was
shown to be capable of killing cells. Over several years researchers
tried to engineer the antibody and succeeded.
In 1997 FDA approved rituximab, now used to treat people with
low-grade lymphoma.
Cancer Vaccines
Cancer vaccines are another form of biological therapy being
studied in the laboratory and in clinical trials. Researchers are
developing vaccines that may promote the recognition of cancer cells
by a person's immune system. These vaccines may help the body reject
tumors and prevent cancer from recurring. In contrast to vaccines
against infectious diseases, cancer vaccines are designed to be
injected after the disease is diagnosed, rather than before it
develops. Vaccines given when the tumor is small may be able to
eradicate the cancer. Cancer vaccines being tested in clinical trials
are designed to treat cancer by getting the immune system to attack
existing cancerous cells. Many vaccines are not used alone, but in
combination with surgery, chemotherapy, or other interventions that
help stimulate the immune response in general.
Early attempts to vaccinate people with cancer against the disease
have been directed largely at melanoma, a potentially deadly skin
cancer with easily accessible tumors. Researchers are also conducting
studies that may lead to the development of vaccines for lymphoma,
prostate, lung, breast, colon, and other cancers.
In the recent past, it has taken 15 years, on average, for an
experimental drug to travel from the laboratory to U.S. consumers.
Often the longest part of the process is finding people to
participate in each clinical trial phase. With increased public
awareness about clinical trials, more people may be willing to
participate, and more professionals may refer people into appropriate
trials. This awareness would ultimately reduce the time it takes for
researchers to enroll participants in trials and complete them--and
speed up the movement of new drugs or treatments into standard
care.
Decisions to Advance Drug Development
Investigators make decisions about how to proceed with further
research based on scientific evidence and promising basic research
leads. Even if some participants in a clinical trial had a positive
response to a new treatment, researchers must look at the global
experience of all participants when deciding whether or not to
continue or expand trials. In some trials, more participants treated
with standard therapy may have better results than those treated with
the experimental therapy, and the investigator may decide to continue
research in a different direction.
|
The
Drug Development and Approval Process in the
1990s
|
|
|
Preclinical Testing
|
Clinical Trials
|
Post-Clinical Trials
|
Total Years for Drug Approval
|
|
|
Step 1
Laboratory / Preclinical
Testing
|
Step 2
File IND1 application with
FDA2
|
Step 3
Phase 1
|
Step 4
Phase 2
|
Step 5
Phase 3
|
Step 6
File NDA3 or BLA4 with
FDA
|
Step 7
FDA Approval
|
 |
|
Purpose
|
Assess safety and biological
activity in the laboratory and in animal
models
|
Obtain FDA approval to begin clinical testing in humans after promising
results in laboratory
|
Determine what dosage is safe,
how treatment should be given
|
Evaluate effectiveness, looks
for side effects
|
Determine whether the new
treatment (or new use of a treatment) is a better
alternative to current standard
|
Inform the FDA of Phase 3 data which supports drug safety and better
performance over standard treatment
|
Review
process/ approval
|
|
All anticancer drugs
(average number of years)
|
4.4 years
|
|
8.6 years
|
|
1.4 years
|
14.4 years
|
|
All drugs*
(average number of years)
|
3.8 years
|
|
10.4 years
|
|
1.5 years
|
15.7 years
|
1IND = Investigational New Drug
2FDA = Food and Drug Administration
3NDA = New Drug Application
4BLA= Biologics License Application
*Classified as "new chemical entities," which exclude
diagnostic agents, vaccines, and other biological
compounds.
Sources: DiMasi, J.A. (2001). New drug development in the
United States 1963-1999. Clinical Pharmacology and
Therapeutics May; 69(5); Tufts Center for the Study of Drugs
Development, Tufts University; adapted from Pharmaceutical
Research and Manufacturers of America.
Refer to the case study 4 for a review and summary of content covered in this workbook.
4. Participant Protection in Clinical Trials
Learning Objectives
Recognize historical events and their influence on
the development of safeguards for participants in clinical
trials
Describe current methods of participant protection
that are implemented throughout the research process
Identify Government regulations and agencies
related to patient protection
|
The strong national and international safeguards in place today to
protect research participants evolved from notorious abuses of human
rights in the past. The first formal statement of medical ethics
regarding research in humans emerged from the 1946 trial and
conviction in Nuremberg, Germany, of Nazi physicians and scientists
who conducted experiments on concentration camp inmates during World
War II. The Nuremberg Code outlined broad concepts for the protection
of human subjects and forms the basis of today's international code
of ethics for the conduct of research.
In the United States, three infamous clinical trials called
attention to the need for participant protection:
The Tuskegee syphilis study, held from 1932 to
1972, followed - but did not treat - poor black men who had
syphilis. During the trial, the men were offered "special free
medical care" and were told that they would be treated for "bad
blood." Instead, more than 400 men with syphilis and 200 men
without the disease who served as controls were enrolled in an
observational clinical trial without their knowledge or consent.
By 1963, it was apparent that many more infected men than controls
had developed complications, and 10 years later a report on the
trial indicated that the death rate among those with syphilis was
roughly double that of the controls. In the 1940s, penicillin was
found to be effective in the treatment of syphilis, but
researchers in the trial, which continued for almost 30 years
after the discovery, neither informed nor treated subjects with
the antibiotic.
From 1963 to 1966, researchers deliberately infected
newly admitted "mentally defective" children at the Willowbrook
School, a State school in New York, with the hepatitis virus in
order to study the natural history of the disease under controlled
circumstances. In some cases, parents were not allowed to admit
children to the institution unless they agreed to let them
participate in the trials.
In 1963, physician-investigators at the Jewish Chronic
Disease Hospital in Brooklyn, New York, injected cancer cells
grown in the lab into people hospitalized with various chronic
diseases without informing the people or gaining their consent. In
review proceedings, the Board of Regents of the State University
of New York found that the trial had not been presented to the
hospital's research committee; the researchers were found guilty
of fraud, deceit, and unprofessional conduct.
In 1974, in response to these tragedies, the President established
the National Commission for the Protection of Human Subjects of
Biomedical and Behavioral Research. In 1979 the commission issued the
Belmont Report, which delineated the ethical principles upon which
today's regulations regarding research participants in the United
States are based:
Respect for persons - recognition of the personal
dignity and autonomy of individuals, as well as special
protections for people with diminished autonomy
Beneficence - the obligation to protect people from harm
by maximizing unanticipated benefits and minimizing possible risk
of harm
Justice - fairness in the distribution of research
benefits and burdens
In addition, the commission concluded that "a permanent board with
the authority to regulate at least all federally supported research
involving human subjects" should be formed.
In response, Congress passed the National Research Act, which
mandated the establishment of IRBs to review all U.S. Department of
Health and Human Services (DHHS)-funded research. The procedures
established for IRBs were further delineated and revised in 1981.
For more details on the history of participant protection, the role of the IRB, and patient confidentiality, see
http://cme.nci.nih.gov.
Two similar sets of regulations - enforced by HHS's Office for
Human Research Protections (OHRP) and FDA - are in place to ensure
the protection of clinical trial participants. If a trial is
Government-supported and it involves an FDA-regulated drug or device,
then it is subject to both sets of regulations. The basic
requirements for IRBs and informed consent are congruent in the two
sets of regulations.
Office for Human Research Protections
The Office for Human Research Protections (OHRP), formerly called
the Office of Protection from Research Risks, safeguards participants
in federally funded research and provides unity and leadership for 17
Federal departments and agencies that carry out research involving
human participants. OHRP enforces an important regulation called the
Common Rule (Title 45 CFR Part 46, Subpart A). The Common Rule sets
standards for:
Informed consent process
Formation and function of IRBs
Involvement of prisoners, children, and other vulnerable
groups in research
Many other protective measures
Researchers must provide written statements describing the
organization of the IRB, its procedures for approving trials, and how
clinical trial participants are protected.
Although breaches in participant protection seldom occur, recent
discoveries of inadequate protection have prompted the restatement of
oversight goals and the addition of some new requirements by OHRP and
the National Institutes of Health (NIH) to strengthen enforcement of
the Common Rule, including:
Aggressive efforts to improve the education and
training of clinical research staff, IRB members, and staff
research administrators regarding protection
Guidelines to reaffirm the need to audit informed
consent records for evidence of full compliance and confirmation
of consent by investigators
Submission of monitoring plans for all phase 1 and 2
clinical trials and the presence of a data and safety monitoring
board for phase 3 trials
Additional information to clarify regulations regarding
conflict of interest
For a detailed proposal of the Government oversight goals set
forth by former HHS Secretary Donna Shalala, see "Protecting Research
Subjects - What Must Be Done" in the September 14, 2000, issue of the
New England Journal of Medicine (343:808-810).
Food and Drug Administration
FDA has its own regulations and policies on IRB review, informed
consent, and participant protection (Title 21 CFR Parts 50 and 56).
The regulations apply to any clinical trial that involves an
investigational drug, biological product, or other device regulated
by FDA, regardless of whether the trial receives Federal funding. FDA
periodically inspects IRB records and operations to certify the
adequacy of approvals, human subject safeguards, and the conduct of
business.
Scientific Review by Sponsor
Clinical trials that are sponsored by NCI are reviewed through
different types of panels, including experts who review the
scientific and technical merit of the proposed research. Many other
clinical trial sponsors, such as pharmaceutical companies, also seek
expert advice on the merits of their studies. Typical issues
addressed by members of expert panels include:
Significance: Does the trial address an important
problem? If its aims are achieved, how will scientific knowledge
be advanced? What effect will the trial have on current concepts
or methods in the field?
Approach: Are the conceptual framework, design, methods,
and analyses adequately developed, well-integrated, and
appropriate? Does the applicant acknowledge potential problem
areas and consider alternative tactics?
Innovation: Does the project employ novel concepts,
approaches, or methods? Are the aims original and innovative? Does
the project challenge existing paradigms or develop new
methodologies or technologies?
Investigator: Do the principal investigator and other
researchers have sufficient training and experience to carry out
the project?
Environment: Does the scientific environment in which
the work will be done contribute to the probability of success? Do
the proposed experiments take advantage of unique features of the
scientific environment or employ useful collaborative
arrangements? Is there evidence of institutional support?
Institutional Review Board (IRB) Approval
An IRB functions as both a clearinghouse and a monitor of clinical
trials. It determines whether the risks involved in a clinical trial
are reasonable with respect to the potential benefits, and it must
approve any clinical trial before it begins. The IRB also monitors
the ongoing progress of the research.
Federal regulations require that an IRB include at least five
people from diverse occupations and backgrounds. In addition, one
member must be outside the sponsoring institution - that is, not
connected to it by employment or relatives. To meet these
requirements, IRBs are usually made up of a mix of medical
specialists, nurses, other health care professionals, ethicists, and
lay members from the community.
Most institutions that carry out clinical trials have their own
review boards (there are roughly 3,000 IRBs in the United States). In
some cases, a small institution might arrange for its research to be
reviewed by another IRB rather than set up its own. All trials that
are federally funded or evaluate a new drug or medical device
regulated by FDA must be submitted to an IRB. However, many
institutions require that all clinical trials conducted in their
facilities, regardless of funding source, be IRB-approved. Before a
trial can begin, the principal investigator submits an application to
an IRB. The board reviews it on the basis of the following
criteria:
Risks to participants are minimized as much as
possible through sound research design
Risks to participants are reasonable in relation to the
anticipated benefits and the knowledge that may result
Participant selection is equitable
Informed consent is sought in accordance with 45 CFR
Part 46.116
Informed consent is documented in accordance with 45 CFR
Part 46.116
Provisions are made for monitoring the data collected to
ensure the safety of participants
Provisions are made to protect the privacy of
participants and the confidentiality of data collected during the
trial
Additional safeguards are in place if any participants
are likely to be vulnerable to coercion or undue influence (e.g.,
children, prisoners, people with mental disabilities, or people
with low income or education levels)
The IRB decides whether to approve the clinical trial and notifies
the researcher and the institution in writing. The IRB may specify
changes the researcher must make in order to gain approval.
After approving a trial, the IRB must decide how frequently to
monitor it - usually on the basis of the risk involved. At the very
least, the trial's progress must be reviewed yearly.
Informed Consent
Informed consent, as a legal, regulatory, and ethical concept, is
an integral part of research. In clinical trials, informed consent is
the process of providing all relevant information about the trial's
purpose, risks, benefits, alternatives, and procedures to a potential
participant, who then, consistent with his or her own interests and
circumstances, makes an informed decision about whether or not to
participate. Before agreeing to take part in a clinical trial,
participants have the right to:
Learn everything that is involved in the trial -
including all details about treatment, tests, and possible risks
and benefits
Both hear and read the information in language they can
understand
Informed Consent Documents
The informed consent form or document provides a summary of the
clinical trial and explains a participant's rights. It is designed to
begin the informed consent process. The participant acknowledges that
he or she is entering a study, has been told what it involves, and
understands the potential risks and benefits of participating.
Although reputable researchers do not try to fool people or sign
them up against their will, individuals sometimes have difficulty
understanding the information about a trial before agreeing to
participate. Individuals may not understand the medical terminology
and/or clinical requirements of a study, and they should be
encouraged to ask questions until they understand all aspects of
treatment. For many people it is important to ask a friend or family
member to come with them when they receive information about medical
options to be sure all important questions are raised. Some people
may want to take notes or bring a tape recorder to assist them with
questions and recall.
The following elements of informed consent are required under the
Common Rule (Title 45 CFR Part 46, Subpart A):
Statement that the trial involves research
Explanation and description of the nature of the trial,
purpose of the trial, duration of participation, procedures to be
followed, and which procedures are experimental
Description of foreseeable risks and discomforts
Benefits to the participant and others
Alternative procedures or treatments
Description of the confidentiality of records
Explanation of procedures if the project involves more
than minimal risk (e.g., compensation, availability of medical
treatment)
Contact person for questions
Statement that participation is voluntary, that there
will be no loss of benefits on withdrawal, and that the
participant may withdraw at any time
Statement that the participant's signature indicates a
decision to participate, having read and discussed the information
presented
Any research trial, regardless of whether it is federally funded,
should provide this information to participants in an informed
consent document.
NCI has issued recommendations designed to help research
institutions and clinical centers write comprehensive, user-friendly
informed consent documents. Its Working Group on Informed Consent
also developed a template and sample forms that serve as models for
covering all of the information that Federal regulations require. To
view the template or other documents related to informed consent, see
the clinical trials section of www.cancer.gov.
Pediatric Assent to Participate
Children and adolescents are not deemed capable of giving true
informed consent, so they are asked for their assent to (or dissent
from) participation in a clinical trial. The trial must be explained
in age-appropriate language or visual aids. Parents or guardians are
asked to give informed permission for their child to participate in a
trial.
Assent must be obtained from all children and young people over
age 7 unless:
The child is found to be incapable of assenting
The clinical trial offers a treatment or procedure that
"holds out a prospect of direct benefit that is important to the
health or well-being of the child and is available only in the
context of the research" (in other words, if the trial offers a
treatment that is thought to be better than those currently
available or if it offers the only alternative to those
available)
Even in these cases, permission from the parent or guardian is
required. For more information, see the
clinical trials section 5 of
www.cancer.gov.
Informed Consent Process
The informed consent process provides people with ongoing
explanations that will help them make educated decisions about
whether to begin or to continue participation in a clinical trial.
The process does not end with the signing of informed consent
documents. If new benefits, risks, or side effects are discovered
during the trial, researchers must inform participants. Participants
are encouraged to ask questions at any time.
Institutional Review Board Role
During the initial review process, the IRB establishes how often a
clinical trial should be monitored. Monitoring occurs at least yearly
but sometimes more frequently. During these review sessions, the IRB
examines a progress report provided by the clinical researcher in
charge of the project. The report describes:
How many people are enrolled in the trial
How many have withdrawn
Participants' experiences, including benefits and
adverse effects
Progress to date
Based on this information, the IRB decides whether the project
should continue as described in the original research plan and, if
not, what changes need to be made. An IRB can decide to suspend or
terminate approval of a clinical trial if the researcher is not
following requirements or if the trial appears to be causing serious
harm to participants.
Data and Safety Monitoring Board (DSMB) Role
NIH requires that all phase 3 clinical trials undergo monitoring
by a DSMB, and that all phase 1 and 2 clinical trials have a data and
safety monitoring plan. A DSMB may also be appropriate and necessary
for phase 1 and 2 clinical trials that are blinded, take place at
multiple clinical sites, or employ particularly high-risk
interventions or vulnerable populations.
The DSMB is an independent committee whose membership includes, at
a minimum, a statistician and a clinical expert in the area being
studied. Other members are experts in all scientific disciplines
needed to interpret the data and ensure participant safety. Members
may also be clinical trial experts, statisticians, bioethicists, or
other clinicians knowledgeable about the trial's subject matter.
The objectives of data and safety monitoring plans are to:
Ensure that risks associated with participation
are minimized to the extent practical and possible
Ensure the integrity of data
Stop a trial if safety concerns arise or if its
objectives are met
Ending Trials Early
There can be compelling reasons for halting a trial early. If
participants experience severe side effects, or if there is clear
evidence that risks outweigh benefits, the IRB and DSMB will
recommend that the trial be stopped early. A trial might also be
stopped if there is clear evidence that the new intervention is
effective - in order to make it widely available.
Breast Cancer Prevention Trial
The Breast Cancer Prevention Trial, conducted by NCI's National
Surgical Adjuvant Breast and Bowel Project, was designed to evaluate
whether taking the drug tamoxifen could prevent breast cancer in
women considered to be at high risk of developing the disease. In
March 1998, interim data showed that tamoxifen cut the chance of
getting breast cancer almost in half. Instead of continuing the trial
for the full 5 years, as planned, researchers stopped the trial after
about 4 years.
Women in the trial who were taking tamoxifen were offered the
opportunity to continue treatment for the remaining 14 months of the
trial. Women receiving the placebo were invited to participate in the
Study of Tamoxifen and Raloxifene, or STAR trial, designed to
determine whether the osteoporosis prevention drug raloxifene is as
effective as tamoxifen in reducing the chance of developing breast
cancer. The women's other option was to seek tamoxifen from a
physician on their own, outside a clinical trial.
B-14 Trial
Another trial involving tamoxifen and conducted by the National
Surgical Adjuvant Breast and Bowel Project, the B-14 trial, was also
halted early - but for a different reason. This trial, which started
in 1982, enrolled women who had had surgery for cancer that was
limited to the breast. After surgery, the women took either tamoxifen
or a placebo for 5 years to determine whether tamoxifen would prevent
recurrence of the cancer. Five years into the trial, significantly
more of the women taking tamoxifen remained disease-free, so the
trial was extended another 5 years. Women who had been taking
tamoxifen were given the opportunity to reenroll in the trial and be
randomly assigned to take tamoxifen or placebo for an additional 5
years.
The extended trial was cut short when several interim data
analyses showed that the tamoxifen group had a slightly higher rate
of cancer recurrence than the placebo group. Statistical analysis
showed that no additional benefit was to be gained by continuing
tamoxifen for more than 5 years. The trial was halted, and the women
stopped taking tamoxifen beyond 5 years.
|
Before taking part in any clinical trial, health care
professionals and their patients should make sure it is
reputable by getting answers to these important
questions:
What is the purpose of the study or therapy?
Who has reviewed and approved it?
What are the credentials of its researchers and
personnel?
What information or results is it based on?
How are study data and patient safety being
monitored?
How will the results be shared?
|
Quality Assurance Monitoring
NCI has several ways of ensuring the quality of data collected
during clinical trials. Many trials, for example, have committees
that review major elements of the study for accuracy, such as:
In addition, data management and statistical centers use quality
control measures to help identify and correct or clarify
inconsistencies and inaccuracies in submitted data.
Another part of NCI's quality assurance program is onsite
monitoring, or audits, of trial procedures, documents, and data.
Institutions are audited at least once every 3 years. Auditors review
three main areas:
Conformance to IRB and informed consent requirements Shipping, storage, and use of drugs and other agents
Individual participants' cases
Adverse Event Reporting
An adverse event is any unanticipated problem involving risks to
clinical trial participants or others. For more information on
adverse event reporting 6,
see cancer.gov/clinicaltrials 5.
Refer to the case study 4 for a review and summary of content covered in this workbook.
5. Barriers to Clinical Trial Participation
Learning Objectives
Compare and contrast benefits and risks of
participating in cancer clinical trials
Identify barriers that deter health care professionals from referring patients to clinical trials
Identify barriers for low participation in cancer clinical trials
Identify barriers that deter special populations
(ethnic minorities, people with limited proficiency in
English, elderly persons) from participating in clinical
trials
Recognize cost and insurance issues related to
participation in clinical trials
|
A 1999 press release from the American Society of Clinical
Oncologists revealed that only 3 percent of adults with cancer
participate in clinical trials - far fewer than the number needed to
answer the most pressing cancer questions quickly.
The reasons so few adults participate in clinical trials are
complex and involve both participant and professional issues. Ideas
to address these problems can be found in
Cancer Clinical Trials: A
Resource Guide for Outreach, Education, and Advocacy 7, also available
from NCI.
Lack of awareness of appropriate clinical trials. Physicians
are not always aware of available clinical trials. Some may not be
aware of the local resources, or some may assume that none would
be appropriate for their patients.
Unwillingness to "lose control" of a person's care. Most
doctors feel that the relationship they have with their patients
is very important. They want what is best for the patient, and if
the person must be referred elsewhere to participate in a trial,
doctors fear they may lose control of the person's care.
Belief that standard therapy is best. Many health care
providers may not adequately understand how clinical trials are
conducted or their importance. Some believe that the treatment in
clinical trials is not as good as the standard treatment. They
also might be uncomfortable admitting that there is uncertainty
about which treatment is best in a phase 3 clinical trial.
Belief that referring and/or participating in a clinical trial
adds an administrative burden. The length and details of most
research protocols may deter providers from participating in
clinical trials. The possibility of incurring additional costs and
expenses that might be inadequately reimbursed is a deterrent for
many.
Concerns about the person's care or how the person will react
to the suggestion of clinical trial participation.
Strategies for Addressing Barriers
- Provide checklists on patient charts with eligibility criteria, placing
posters with open protocols listed, or using abridged "protocol
pocket cards" with key inclusion and exclusion criteria.
- Dedicate one research nurse or research assistant to identifying and
screening participants, coordinating pre-enrollment tests, educating
participants about the protocol and process, and initiating the
informed consent and enrollment process.
- Access funding for clinical trial support. See the www.cancer.gov site
section on clinical trials, the CTSU site (www.ctsu.org), and the NCI
Cooperative Group sites for information.
Lack of awareness of clinical trials. Research has
consistently shown that most people with cancer are not aware of
the option to participate in clinical trials.
Lack of access to trials. The reality or the perception that
there are no trials nearby deters many potential participants. In
addition, seeking care at a distant trial site presents time and
travel barriers.
Fear, distrust, or suspicions of research. For many people,
the loss of control (not choosing their treatment) that comes with
entering a randomized trial is too great. Many also fear being
treated like "guinea pigs" or being "experimented upon," as well
as not receiving treatment for their cancer. People may have a
general lack of trust in the medical profession based on past
negative experiences or the knowledge of historical abuses of
research participants.
Practical or personal obstacles. Costs of being away from work
and family may be deterrents for some people. Others may not wish
to leave the care of their own physician. People from certain
racial or ethnic groups or who are medically underserved may feel
that care within a trial will not be sensitive to their needs.
Others may feel that recruitment strategies are not sensitive to
their needs. Still others may believe that standard care is better
than the treatment available in a trial.
Insurance or cost problems. Another deterrent is the fear of
being denied insurance coverage for participation in a clinical
trial. If a person is uninsured, the cost of trial participation
is an issue.
Unwillingness to go against personal physician's wishes.
|
A Survey on Clinical Trial Barriers
A survey of almost 6,000 people with cancer conducted in
2000 took a look at why so few adults participate in cancer
clinical trials. Some of the highlights included:
About 85 percent of people with cancer were either
unaware or unsure that participation in clinical trials was
an option, though about 75 percent of these people said they
would have been willing to enroll had they known it was
possible.
Of those who were aware of the clinical trial
option, most declined to participate because they believed
common myths about clinical trials. They either thought
that:
The medical treatment they would receive in a
clinical trial would be less effective than standard care
They might get a placebo
They would be treated like a "guinea pig"
Their insurance company would not cover costs
People who received treatment through a clinical
trial found it to be a very positive experience:
Ninety-seven percent said they were treated
with dignity and respect and that the quality of care
they received was "excellent" or "good"
Eighty-six percent said their treatment was covered
by insurance
Source: Harris Interactive. Health Care News 1(3)
[Poll].
(Available from
www.harrisinteractive.com/harris_poll/ 8
Supported by the Coalition of National Cancer Cooperative
Groups, the Cancer Research Foundation of America, the
Cancer Leadership Council, and the Oncology Nursing
Society.
|
Additional barriers exist for people who are from certain
ethnic/racial backgrounds or who are medically underserved. The
following is not meant to be a comprehensive overview of all barriers
associated with clinical trials, and what is outlined should not be
generalized to all diverse populations.
Diverse U.S. Populations - Definitions
Diverse populations include minority ethnic and racial groups
designated by the U.S. Government, including:
American Indian or Alaska Native
Asian American
Black or African American
Hispanic or Latin American
Native Hawaiian or other Pacific Islander
Ethnically diverse populations are growing rapidly; in the 2000
Census, about 25 percent of the U.S. population reported their race
as something other than White.
The National Cancer Institute's working definition of diverse
populations also includes medically underserved populations.
Medically underserved populations are those that lack easy access to,
or don't make use of, high-quality cancer prevention, screening and
early detection, treatment, or rehabilitation services. These may
include people of any racial or ethnic group who live in rural areas,
or who have low income or literacy levels. Medically underserved
groups are generally characterized as experiencing higher cancer
mortality rates and insufficient participation rates in cancer
control programs.
Specific Barriers
Long-standing fear, apprehension, and skepticism exist among
some minority populations about medical research because of abuses
that have happened in the past (e.g., the legacy of the Tuskegee
syphilis study). Among these populations, there is often
widespread fear and distrust of the medical care system as a
result of discrimination, indifference, and disrespect. Many feel
that they do not want to give up rights or lose power in order to
be "experimented on." Others may be skeptical about the quality of
care that would be provided in a clinical trial. Some may find
that trial recruitment strategies are not sensitive to their
needs.
Doctors may not mention clinical trials as an option for
cancer care. As noted above, many physicians do not refer people
to clinical trials. Some physicians may avoid suggesting a
clinical trial to people who belong to racial or ethnic minorities
out of concern that people would see them as insensitive.
Moreover, some physicians may inadvertently discriminate against
older people or those from certain ethnic or cultural
backgrounds.
People from various cultural or ethnic backgrounds hold
different values and beliefs that may be different than principles
of Western medicine. Many people have cultural beliefs that
Western medicine cannot address their health concerns. Different
ethnic and cultural views of health and disease (e.g., fatalism,
family decisions about treatment, use of "traditional healers,"
prayer, herbal medicines, or use of complementary/alternative
health practices) may make clinical trials a less attractive
treatment option. For prevention trials, many may feel that the
risk of a potential disease and its consequences may be less
important than meeting daily needs.
Language or literacy barriers may make it difficult for some
people to understand and consider participating. The complexity of
forms, including informed consent documents, may also be a barrier
to those considering participation. Translation can also be
difficult if the person translating information has not had
specialized training.
Additional access problems confront many people. Depending on
where they live or their access to transportation, people may have
difficulty getting to a clinical trial site. Those with low
incomes may find it difficult to take time off work or find
appropriate childcare. Other barriers, such as a lack of health
insurance or a source of health care, clearly present difficulties
in accessing trials.
For some solutions to barriers for diverse populations, see NCI's Cancer Clinical Trials: A Resource Guide for Advocacy, Education, and Outreach. 7
The costs associated with clinical trials can be a barrier for
many professionals and the public. Physicians are often concerned
about reimbursement related to the expense of either caring for
people enrolled in trials or offering trials within their practice.
Potential trial participants often fear that their insurance company
will not cover participation. Those who are uninsured will need to
know how their participation in a trial will be covered.
There are two types of costs associated with clinical trials:
participant care costs and research costs.
Participant Care Costs
Participant care costs include:
-
Usual care costs, such as doctor visits, hospital
stays, clinical laboratory tests, and x-rays, occur whether
someone is participating in a trial or receiving standard
treatment.
Extra care costs are those associated with clinical
trial participation, such as additional tests that may be
required.
These costs may or may not be covered by a participant's health
plan.
Research Costs
Research costs include costs associated with conducting the trial,
such as:
Data collection and management
Research physician and nurse time
Analysis of results
Clinical laboratory tests and x-rays
Cost of the agent being tested
Most of the time, research costs are covered by the sponsoring
organization.
Health Plan Coverage
Health insurance companies and managed care plans do not always
cover all care costs in a clinical trial. What they do cover varies
by plan and by trial. Now that Medicare has developed a policy
explicitly covering the routine care costs of diagnostic and
treatment clinical trials, other insurers may follow suit.
Insurance companies often claim that paying for clinical trials
would be too costly. But recent studies (Bennet et al., 2000; Fireman
et al., 2000; Wagner et al., 1999) found that costs for participants
in clinical trials are not appreciably higher than costs for people
not enrolled in trials.
For coverage strategies for participants and professionals, see
the clinical trials section of www.cancer.gov. Some insurance
carriers that cover clinical trials will even help health care
professionals locate appropriate trials.
Established vs. Investigational Therapies
In general, the most important factor determining coverage of a
treatment is the health plan's judgment as to whether the therapy is
"established" or "investigational." Health plans usually consider a
treatment established if sufficient scientific data exist to show it
is safe and effective. If a health plan does not think sufficient
data exist, it may consider the service investigational. Thus, some
health plans, especially smaller ones, will not cover any costs
associated with a clinical trial. Policies vary widely, but in most
cases it helps to have someone from the research team initiate
discussions with the health plan.
Other Criteria
Health plans may specify other criteria a trial must meet in order
to be covered, for example:
Sponsorship: The trial must be sponsored by an
organization whose review and oversight procedures meet the health
plan's standards of scientific rigor.
Trial type and phase: The trial must be judged
"medically necessary" by the health plan; this determination is
made on a case-by-case basis. In some cases, the trial must be in
phase 3. While a plan may be willing to cover costs associated
with phase 3 trials, it may require documentation of known
benefits before covering a phase 1 or 2 trial. Participants may
have more difficulty getting coverage for costs associated with
prevention and screening trials because health plans are currently
less likely to have a review process in place for them.
Cost neutrality: The trial must be cost-neutral - that
is, it must not be significantly more expensive than treatments
the health plan considers standard.
Lack of standard therapy: The trial must offer treatment
of a cancer for which no standard therapy is available.
Facility and personnel qualifications: The facility and
medical staff must meet specific health plan qualifications for
conducting unusual services, especially intensive therapy such as
a bone marrow transplant (high-dose chemotherapy with bone
marrow/stem cell rescue).
Legislation and Policies
Despite interest at the Federal level, as of 2002, no legislation
has been passed to require private health plans to uniformly cover
all clinical trial costs. However, there have been several important
developments at the Federal level regarding clinical trial
coverage:
Medicare reimburses for all routine participant
care costs for its beneficiaries participating in clinical trials.
Beneficiaries of TRICARE, the Department of Defense's
health program, are covered for NCI-sponsored phase 2 and phase 3
prevention and treatment clinical trials.
The Department of Veterans Affairs (VA) allows eligible
veterans to participate in a broad range of NCI clinical trials
across the country. The agreement covers all phases and types of
NCI-sponsored trials.
Many States have also passed legislation or developed policies
that require health plans to cover clinical trial costs. For an
updated
legislation listing 9,
see the clinical trials 5 section of the
NCI Web site cancer.gov 10.
Refer to the case study 4 for a review and summary of content covered in this workbook.
6. Conducting, Referring to, and Locating Clinical Trials
Describe the types of sponsorship of cancer
clinical trials
Define the role of the National Cancer Institute
in conducting clinical trials throughout the United
States
Identify methods of referring patients to clinical
trials
Demonstrate ways of locating clinical trial
resources
|
In order to find a clinical trial, it is helpful to understand who
sponsors trials. Clinical trials are sponsored by both Government and
private organizations including the National Cancer Institute and
pharmaceutical companies. Clinical trials take place all over the
United States, in locations as diverse as a rural community clinic or
a cancer center in a large urban area.
National Cancer Institute
NCI sponsors hundreds of clinical trials around the country
through six major programs, which are discussed below.
Clinical Trials Cooperative Group Program
The Clinical Trials Cooperative Group Program supports a large
network of organizations that continually generate and conduct new
clinical trials consistent with national priorities in cancer
treatment research. Member organizations carry out large, randomized
phase 3 trials, as well as phase 2 trials.
Members conduct clinical trials in locations nationwide, while
administration and data are handled at a central location. Members of
the groups include:
Academic institutions
NCI-designated cancer centers
Physicians in the Community Clinical Oncology Program
and the Minority-Based Community Clinical Oncology Program
(described below)
Community physicians and community hospitals
Each year, the groups enroll some 20,000 new people in treatment
trials, evaluate some 12,000 new participants in ancillary studies,
and follow the progress of many times that many participants in
ongoing trials. Thousands of individual investigators participate in
these studies. The cooperative groups have been instrumental in
developing both new standards of care for people with cancer and
sophisticated clinical investigation techniques.
There are 12 groups in the Clinical Trials Cooperative Group Program:
American College of Surgeons Oncology Group (ACOSOG)
Cancer and Acute Leukemia Group B (CALBG)
Children's Cancer Study Group (CCSG)
Eastern Cooperative Oncology Group (ECOG)
Gynecologic Oncology Group (GOG)
Intergroup Rhabdomyosarcoma Study Group (IRSG)
National Surgical Adjuvant Breast and Bowel Project (NSABP)
National Wilms Tumor Study Group (NWTSG)
North Central Cancer Treatment Group (NCCTG)
Pediatric Oncology Group (POG)
Radiation Therapy Oncology Group (RTOG)
Southwest Oncology Group (SWOG)
For more cooperative group information, see http://ctep.info.nih.gov.
Select "Cooperative Group Program."
Cancer Trials Support Unit
NCI and the Clinical Trials Cooperative Group Program are
collaborating in a pilot project - the Cancer Trials Support Unit
(CTSU) - to reduce administrative burdens and recruit more physicians
to become involved in clinical trials.
CTSU is designed to streamline and centralize many administrative,
financial, and data collection tasks. CTSU will provide participating
physicians with a single access point to NCI's entire phase 3
clinical trial system, facilitating access to protocols, training,
and educational information. Highlights of the fully developed system
will include:
Cross-group registration enabling physicians to
register people on adult cooperative group trials in leukemia,
lung, genitourinary, colorectal, and breast cancers, even if they
are not members of the group conducting the trial
Online registration, eligibility assessment, and
reporting of data that will use a common format and
state-of-the-art data management systems
A coordinated auditing system that will eliminate
multiple quality assurance audits for research personnel
participating in more than one cooperative group
Centralization of administrative tasks, including
credentialing and verification of IRB approval for all
investigators participating in cooperative groups and CTSU
CTSU opened in July 2000 for cooperative group members. If the
initial experience is successful, oncologists not affiliated with a
group will be encouraged to participate as well.
For more information about CTSU, see www.ctsu.org.
Community Clinical Oncology Program
The Community Clinical Oncology Program (CCOP) enables community
physicians to work with investigators conducting NCI-supported
clinical trials. The program increases the number of participants and
physicians who can take part in clinical trials operated
simultaneously in major research centers and in the community. It
benefits investigators by giving them an opportunity to conduct
large-scale cancer prevention and control studies at the community
level.
Facilities participating in the program must be affiliated with an
NCI-supported clinical cooperative group or cancer center and use
research protocols developed by these groups.
Minority-Based Community Clinical Oncology Program
The Minority-Based Community Clinical Oncology Program (MBCCOP)
was initiated in 1990 to provide people with cancer who belong to
minority groups access to state-of-the-art treatment, prevention, and
control technology. The minority-based program was begun because 40
percent of the people with cancer referred to CCOP physicians each
year are from a minority group. Each MBCCOP pledges to accrue more
participants from minority groups than other CCOPs do.
Since funding began, participant enrollment in the minority-based
program has grown to account for approximately 10 percent of all
ethnic minorities enrolled in NCI-approved clinical trials.
Cancer Centers Program
The Cancer Centers Program consists of more than 50 NCI-supported
research centers. Each cancer center also belongs to at least one
cooperative group.
NCI supports three types of centers:
Comprehensive cancer centers, which conduct basic, clinical,
and preventive research programs, as well as community outreach and
education programs
Clinical cancer centers, which conduct primarily clinical
research programs but may have programs in other research areas as
well
Cancer centers (formerly called Basic Science Cancer Centers),
which conduct basic or preventive research programs and do not have
clinical programs
Please refer to http://cancercenters.cancer.gov/cancer_centers/index.html
for a comprehensive list of cancer centers.
Clinical Grants Program
The Clinical Grants Program supports clinical researchers through
various types of grants, all of which are peer reviewed.
Pharmaceutical and Biotech Companies
Pharmaceutical and biotech companies also conduct clinical trials,
both locally and nationally. They may conduct these trials in
collaboration with universities, hospitals, NCI, and local
physicians. These trials are subject to the companies' own review
panels and to an IRB, which may be local or national in scope.
When a person is first diagnosed with cancer, his or her physician
may suggest several possible options for treatment, possibly
including a clinical trial. Likewise, a person at high risk of
developing cancer may be offered prevention options that include a
clinical trial.
Decisions concerning eligibility for cancer clinical trials are
often complicated, requiring very specific information regarding a
person's medical condition and prior treatment. For that reason, it
is preferable to have a provider who is familiar with the person's
case make the initial contact with clinical trial staff. A person who
calls a researcher directly may have insufficient medical
information, thereby making a decision about eligibility difficult
and frustrating for both the potential participant and the
researcher. Once contact is made, a referral coordinator may accept
telephone, mail, or e-mail inquiries from physicians, potential
participants, and others about the availability of clinical trials.
Preliminary eligibility can be evaluated over the phone, and
appointments with the clinical trial team can be scheduled if the
person decides to proceed.
The informed consent process begins as soon as a potential
participant begins to explore the trial with the clinical research
team. The research team discusses the trial's purpose, procedures,
risks, potential benefits, and participants' rights. After this
dialogue, the person may wish to speak to his or her referring
provider to review the information and help determine the best
course.
If the person opts to enter the trial, the research team should
send the referring provider relevant updates and followup information
until the person returns to the provider's care.
Deciding to refer a person to a clinical trial is easier if the
health care professional has adequate information regarding the
trial's objectives and eligibility criteria. Before discussing a
trial as an option, the professional should learn as much as possible
about the trial.
Discussing Clinical Trials with Patients
Health care professionals may be able to assist potential trial
participants in their decision by considering the following benefits
and risks to clinical trial participation with their patients.
Possible Benefits of Clinical Trial Participation
Participants will receive, at a minimum, the best
standard treatment. This may be as good as, or better than, the
new approach being tested.
If a participant is taking the new treatment and it is
shown to work, he or she may be among the first to benefit.
By examining the benefits and drawbacks of clinical
trials and other treatment choices, participants are taking an
active role in a decision that affects their health care and
life.
Some participants feel good about helping advance
medical knowledge that will improve cancer care and help
others.
Even when they don't lead to new therapies, clinical
trials often answer important questions and help move research
forward.
Possible Risks of Clinical Trial Participation
New treatments are not always better than standard
care.
New treatments may have unexpected side effects that are
worse than those of standard treatment.
Although a new treatment is beneficial, it may not work
for every participant. Even standard treatments do not help
everyone.
If a participant receives standard treatment instead of
the new treatment being tested, it may not be as effective as the
new approach.
Participants may have additional patient care costs that
are not covered by the study sponsor or by their health insurance
or managed care plan.
Participants may have to incur the costs of travel,
childcare, lost work hours, hotels, and meals.
Participation in a clinical trial may require increased
patient responsibility, such as going to more appointments or
self-monitoring for side effects.
NCI Resources
NCI's Web site, Cancer.gov 10,
provides access to a wealth of information on clinical cancer care.
The site contains information from PDQ® (Physician Data Query), including the latest
information about cancer treatment, screening, prevention, genetics,
supportive care, and complementary and alternative medicine, as well
as a registry of cancer clinical trials. Clinical oncology
specialists review current literature from more than 70 medical
journals, evaluate its relevance, and synthesize it into clear
summaries, which are then reviewed monthly and updated as needed based on new
information. Most cancer information summaries appear in two
versions: 1) a technical version for the health professional and 2) a
nontechnical version for patients, their families, and the public.
Many of the summaries are also available in Spanish.
The NCI Web site also includes approximately 100 fact sheets on
various cancer-related topics, information on ordering NCI
publications, and educational features and news summaries concerning
the latest results from cancer clinical trials.
NCI also has a Web-based continuing education tutorial on implementing clinical trials into your practice.
This tutorial provides information for health care professionals interested in referring patients to trials, and more detailed information for health care professionals interested in actually implementing clincial trials into their practice. The series is available at cme.cancer.gov 11.
The clinical trials registry PDQ contains more than 1,800 ongoing
clinical trials, with information about trials around the world. All
clinical trials undergo review prior to inclusion. Although no single
resource lists every cancer clinical trial being conducted in the
United States and abroad, PDQ is the most comprehensive cancer
clinical trials registry, and contains information about trials
sponsored by NCI, the pharmaceutical industry, and some international
groups. Users can narrow their search by multiple parameters, such as
stage of disease, phase of trial, treatment modality, and geographic
location. PDQ also contains an archival file of more than 11,000
clinical trials that are no longer accepting participants, including
contact information for principal investigators of trials that may
not yet be published in the biomedical literature.
Accessing NCI's Clinical Trial and Cancer Information by Phone or
Fax
NCI's Cancer Information Service 12 - NCI's Cancer Information
Service is a national information and education network for patients,
the public, and health professionals. From regional offices covering
the entire United States, Puerto Rico, and the U.S. Virgin Islands,
trained staff provide the latest cancer information through a
toll-free telephone service. Staff can respond to calls in either
English or Spanish. The Cancer Information Service, with regional offices throughout the United States, may work with organizations and professionals to plan, implement, and evaluate culturally appropriate clinical trials education programs using the Clinical Trials Education Series. 13
Access: The toll-free number is 1-800-4-CANCER
(1-800-422-6237). For deaf and hard of hearing callers with TTY
equipment, the number is 1-800-332-8615. Hours of operation are
Monday through Friday, 9:00 a.m. to 4:30 p.m., local time. Callers
also have the option of listening to recorded information about
cancer 24 hours a day, 7 days a week.
NIH Web Site
No single resource lists every cancer clinical trial being
conducted in the United States and abroad. However, in 2000 the
National Institutes of Health launched a new Web site,
www.clinicaltrials.gov,
that aims to be a complete listing of all
U.S. Government- and industry-sponsored clinical trials, including
cancer trials.
The site contains approximately 7,200 clinical trials, most of
them Government-sponsored. However, additional trials from the
pharmaceutical industry are being added.
Other Web Sites
The Internet includes a variety of clinical trial databases and
matching services. The owners of these sites can be:
Not-for-profit organizations, that might:
For-profit organizations, that might:
Anyone interested in using online services to find a clinical
trial should ask questions and evaluate the information before
submitting personal information or calling an investigator from the
service:
Who owns/runs the site?
Where does the financial backing come from?
How does the service get paid? By matching people to
trials? By clinical trial submission to the database? Other?
Does anyone make money on this site? If so, who?
What is the source of clinical trial information?
Does the site include all clinical trials? All
Government-supported trials? All pharmaceutical trials?
People may wish to look at information from many sites and
consider the source of the information before making important
health-related decisions.
|
Guide To
Finding Clinical Trial Resources
|
|
National Cancer Institute's PDQ
|
What is it?
|
How do I access it?
|
What will it provide?
|
|
Database produced by NCI
Registry of approximately 1,800 active cancer clinical
trials
|
Cancer.gov 10
Go to the clinical trials area and follow the search
directions OR Call 1-800-4-CANCER
|
Summaries about clinical trials conducted by
NCI-sponsored researchers, the pharmaceutical industry, and
some international groups
|
|
National Library of Medicine
|
Database produced by NIH
Registry now lists 4,000
primarily NIH-supported clinical studies on many conditions,
and more will be added
All trials on PDQ are listed in this database
|
Go to www.clinicaltrials.gov
Can browse by disease or sponsor or insert key words
|
Summaries about clinical trials for a wide range of
conditions - most of the trials listed are sponsored by
NIH
|
|
Local Cancer Center Web Sites
|
Locally produced Web sites that include listings for
trials sponsored by NCI and some pharmaceutical
companies
Good supplementary resources for locating clinical
trials; a cancer center may begin participating in an
NCI-sponsored trial before the center's information is
listed in PDQ
|
Different sites can be found through:
Information on trials taking place at NCI's Clinical
Center in Bethesda, Maryland is available at http://ccr.nci.nih.gov then select "clinical trials"
Some centers may also have telephone information
centers
|
Information that varies from center to center
|
|
Example of Pharmaceutical Resources/Internet Clinical
Trial Matching Sites
|
Pharmaceutical Research and Manufacturers of America
(PhRMA) publishes a list of new cancer drugs in
development
CenterWatch's Clinical Trials Listing Service and EmergingMed.com's clinical trials matching service list many
industry- and Government-sponsored trials
|
PhRma.org 14
Click on "New Medicines in Development" and search by
disease. The drugs are listed by cancer type. Or call 202-835-3400.
CenterWatch.com 15
Click on "Trial Listings" and then "CenterWatch Trial
Listings by Medical Areas" or call 617-856-5900
EmergingMed.com 16 or call 877-601-8601
|
Descriptions, sites, telephone numbers, and investigator
names by state
|
Refer to the case study 4 for a review and summary of content covered in this workbook.
7. Case Study
Mr. Joe Smith, a 59-year-old African American male, presents to
his primary care physician, Dr. Bob Brown, in rural Wisconsin. He has
been complaining of difficulty with initiating urination and
frequency. He presents clinically with an abnormal rectal exam and an
elevated prostate-specific antigen (PSA) of 8 ng/ml (normal in a
59-year-old Black male is less than 4 ng/ml). Mr. Smith's mass is
biopsied and the results reveal a prostatic tumor. The nearest
comprehensive cancer center is 100 miles away, and Mr. Smith wants to
be treated by Dr. Brown. He says he wants to stay close to his home
and family, and does not want to be cared for by "those big city
doctors."
Mr. Smith decides to undergo a radical prostatectomy at his
community hospital. Surgery reveals a 1.5 cm tumor with clear
margins, negative pelvic node involvement, and unilateral seminal
vesicle involvement, which puts Mr. Smith at high risk for eventual
tumor spread. To complete his clinical staging, a bone scan and CT
scan are completed. Mr. Smith's tumor is formally staged. At his
3-month followup visit, post-operatively, his PSA is 0.2
(undetectable).
Dr. Brown closely follows Mr. Smith. Three years after his surgery
his PSA has slowly risen to 11, but his bone scan and CT scans remain
negative. Dr. Brown informs Mr. Smith that there are many clinical
trials for men with prostate cancer at all stages. He explains that
these trials are being conducted to try to find the best methods for
cancer prevention, early detection, and treatment. At this point in
his disease, Mr. Smith does not wish to consider a clinical trial, so
he continues to receive standard care. Dr. Brown recommends starting
standard treatment with hormone therapy injections monthly. The
patient's PSA drops to less than 0.2 again.
Two years later, the PSA begins to rise to 15 and a bone scan now
shows abnormal uptake in multiple ribs and the thoracic spine. Mr.
Smith is started on an anti-androgen drug and his PSA drops to 10 at
his 3-month followup visit. One year later his PSA has risen to 40,
and he now says he has mild rib pain. Dr. Brown explains to Mr. Smith
that he needs to consult with an oncologist from the cancer center,
Dr. Mary Jones, and that Mr. Smith may need to see her. Mr. Smith
somewhat reluctantly agrees to see Dr. Jones. Dr. Jones recommends
exploring available clinical trials because Mr. Smith is young, is in
good health with mild symptoms, and has a tumor that now appears to
be progressing.
How can Dr. Brown or Dr. Jones find an appropriate clinical trial
for Mr. Smith?
Dr. Jones decides to check NCI's PDQ system, because her center
has no active trials for prostate cancer. The PDQ system will allow
her to see current active clinical trials and their eligibility
criteria. (More information on PDQ 17.)
Dr. Jones uses the NCI PDQ system to assess what, if any, clinical
trials are available for Mr. Smith. Using the Internet, she accesses
the site as follows:
Enters the clinical trials section of Cancer.gov 10
Selects "Finding Clinical Trials"
Selects PDQ Search Form
Enters the relevant data; hits search (appropriate
studies will be retrieved)
Reviews the trials
If a physician does not have Internet access, how can he or she
find a trial?
Clinical trial information is always available through NCI's
Cancer Information Service (CIS) at 1-800-4-CANCER
(1-800-422-6237).
After reviewing available trials, Dr. Jones selects a phase 2
trial entitled: A Phase II Randomized Study of High-Dose Ketoconazole
With or Without Alendronate Sodium in Patients With Androgen
Independent Metastatic Adenocarcinoma of the Prostate (2001) to
discuss with Dr. Brown and Mr. Smith.
Mr. Smith is clinically eligible for the study. Participants are
randomized to one of two arms: 1) a single oral dose of ketoconazole
on day 1, and then 3 times daily oral dose beginning on day 8, versus
2) a single oral dose of alendronate sodium on day 1 and a single
oral dose of ketoconazole on day 3 and then daily alendronate sodium
and 3 times daily ketoconazole beginning on day 8. Participants who
experience a clinically complete remission (CR) receive treatment for
an additional 60 days beyond CR. Mr. Smith would have to be willing
to have followup visits at NIH in Bethesda, MD, every 2 months and be
seen in the NIH Cancer Center every 2 weeks for evaluation of
toxicity and drug tolerance.
Randomization
As a health care professional it is often challenging to discuss
the concept of randomization with patients. Patients can feel
threatened knowing that neither they, nor their doctor, can choose
what treatment they will get if they enter a randomized trial. They
may feel that the arm of the study that is standard care or closest
to standard care, is the best option. People may also be concerned
that if they enter a randomized clinical trial they will not receive
treatment; they may receive just a "sugar pill." How can they be
sure?
Dr. Jones began her discussion of randomization with Mr. Smith,
his wife, and their two grown sons by explaining that this study has
an objective group of professionals who monitor the study called a
data and safety monitoring board (DMSB). The DMSB evaluates the data
from the clinical trial to interpret if the therapy or technique
being studied appears to be better (more beneficial) or worse (more
harmful) than standard therapy. A trial can be halted early to allow
all participants access to a clearly more beneficial intervention, or
to protect participants from harm.
Dr. Jones explained that there are three phases for clinical
trials and that randomization is generally seen in phase 3 studies
where researchers are unsure whether a new intervention is better
than the currently accepted standard therapy for a specific cancer
type. She discussed how patients entering a phase 3 trial (and, as in
this case, some phase 2 trials) are randomly assigned to groups
(called randomization) and that neither the patients nor their
doctors choose which therapy or technique they will receive. This is
done because if physicians determined which therapy participants
would receive, there could be an unconscious bias in their
assignments. They could tend to assign patients with a more hopeful
prognosis to the experimental therapy group and make the new therapy
seem more effective than it really is. Similarly, if patients were
allowed to choose which therapy they would receive, the results could
also be influenced. Patients with a less hopeful prognosis could tend
to pick the experimental treatment, for example, which may lead that
treatment to look less effective than it really is.
Myths
Mr. Smith and his family have many questions regarding what they
have heard and read about clinical trials. How would you answer these
questions?
1. How do I know I am really safe if I enter this trial? You and I
both know there are bad feelings in our community about research.
Fact: Tragedies have occurred in the past related to
clinical trials. The African American community most notably
remembers the infamous Tuskegee syphilis study, which followed,
but did not treat, African American men with syphilis. There are
now strong safeguards in place to protect research participants
from the notorious human rights abuses of the past which include:
Government oversight and regulations
Institutional review board (IRB) review and approval
of a clinical trial before it begins and annually while it is
in progress
An informed consent process which gives potential
participants the information they need to decide about
participation, including foreseeable risks and benefits
Data and safety monitoring boards which ensure
minimization of risks, integrity of trial data, and oversight
to end a trial early if clear benefit or harm arises from the
intervention being studied (usually used in phase 3
studies)
2. Aren't these clinical trials only for dying cancer
patients?
Fact: Clinical trials are not just for patients with the
most advanced disease. In fact, many newly diagnosed cancer
patients participate in clinical trials. If only the sickest
patients participated in treatment trials, researchers would not
know how to treat patients with earlier stages of cancer. Phase 3
treatment includes all stages of cancer, from the most advanced to
the most localized. These trials enroll hundreds or thousands of
patients. Phase 1 and 2 cancer clinical trials, which enroll fewer
than 100 patients, seek people with few treatment options or
people who have exhausted all the current treatment options, which
is Mr. Smith's case.
3. Aren't people who join clinical trials just "guinea pigs" for
research?
Fact: People who decide to take part in a clinical trial
are called participants, and strict guidelines are in place to
ensure that these volunteers are treated as such:
A participant has the right to withdraw from a
trial at any time. The participant's decision does not
jeopardize his or her future treatment and he or she may
discuss further treatment options with the study physician or
be referred back to a primary care provider for standard care.
Although people fear that trial participants are
treated like guinea pigs, reports from actual trial
participants disagree. According to a Harris Poll conducted in
2000, the vast majority of trial participants said their
overall experience was positive. Ninety-seven percent said they
were treated with dignity and respect and that the quality of
care they received was "excellent" or "good." More than 80
percent said they did not receive more tests than they felt
were necessary and 86 percent said their treatment was covered
by insurance.
4. Mr. Smith's wife is very concerned that cancer patients who
join clinical treatment trials get a sugar pill (placebo) instead of
really being treated. Will my husband really get treatment?
Fact: In phase 3 cancer treatment trials, participants
with cancer get either a new treatment or the best standard
treatment. In phase 2 trials, like the one Mr. Smith is
considering, different schedules and combinations of two drugs are
being evaluated for their effectiveness. It is unethical to
deprive any person with a serious illness or condition of the best
available treatment. There would be no placebo involved in Mr.
Smith's treatment.
5. His oldest son is wondering if only people who have cancer can
participate in a clinical trial. Aren't my brother and I at higher
risk now?
Fact: Treatment and diagnostic trials are designed for
people who already have cancer; however, genetics, prevention, and
screening trials are designed for persons at risk of developing
cancer. This is relevant for Mr. Smith's sons who now have an
increased risk for prostate cancer. Dr. Jones suggests that the
sons make an appointment to discuss a cancer prevention clinical
trial.
Insurance
Dr. Brown is wondering whether participating in this cancer
clinical trial will cost Mr. Smith more than standard treatment. Will
insurance cover the costs?
The costs of new treatments for different cancers vary. Some
treatments under study cost no more than standard therapies. Others,
such as bone marrow transplants, are very expensive. There are
hundreds of insurance companies and managed care organizations in the
United States and each has a different policy about covering clinical
trial costs. In general, most companies have contract language that
prohibits coverage for "experimental therapies." However, decisions
are often made on a case-by-case basis, and costs for patient care in
clinical trials are often covered.
The best way to evaluate each situation is to ask questions such
as the following:
What will the total patient care costs be?
What parts of the treatment, if any, does the study
provide free of charge?
What parts of treatment must be paid for by the
participant or the participant's insurer?
What is the situation for people who have no health
insurance?
Will total patient charges be higher for a clinical
trial
than for standard care?
How often have insurers reimbursed all costs of the new
therapy?
Are there other resources or organizations that might
help cover the fees or provide services, such as free
transportation?
Another option is to discuss reimbursement issues with the
insurance company ahead of time. The company is unlikely to promise
coverage before the fact, but it may give information about general
policies and trends. In considering costs of out-of-town treatment or
follow-up care, patients should remember to include travel-related
costs. The research team conducting the study will know how many
times participants will need to visit, for how long, whether housing
or stipends will be provided, and whether participants will be
hospitalized during their stay.
After meeting with Dr. Jones and discussing their concerns, the
Smith family met with Dr. Brown to discuss his view of the trial. Mr.
Smith decided to enter the trial. After four months and his second
evaluation, his PSA has decreased to 14 ng/ml, and he has no bone
pain. He will continue for another cycle of therapy and be
reevaluated in 2 months. After his initial apprehension eased and
treatment began, Mr. Smith was able to verbalize satisfaction in both
his decision to enter the trial and in the level of care and
attention he received.
Glossary
adjuvant therapy: One or more anticancer drugs used in
combination with surgery or radiation therapy as part of the
treatment of cancer. Adjuvant therapy is given before or after the
primary treatment to increase the chances of a cure. Adjuvant usually
means "in addition to" initial treatment.
adverse effect: See side effects.
Adverse Event Expedited Reporting System: A Web-based
program that enables researchers using NCI-sponsored investigational
agents to expedite the reporting of serious and/or unexpected adverse
events directly to NCI and FDA.
agent: In a cancer clinical trial, an agent is a substance
that researchers believe might be capable of producing an effect that
fights cancer.
assent: Children and adolescents are not deemed capable of
giving true informed consent, so they are asked for their assent, or
agreement, to participation in a clinical trial.
audit: In clinical trials, the onsite monitoring of trial
procedures, documents, and data.
Belmont Report: A 1979 report by the National Commission
for the Protection of Human Subjects of Biomedical and Behavioral
Research that delineated the ethical principles upon which today's
regulations regarding research participants in the United States are
based: respect for persons, beneficence, and justice.
bias: Human choices, beliefs, or any other factors besides
those being studied that affect a clinical trial's results. Clinical
trials use many methods to avoid bias because biased results may not
be correct.
biological therapy: Treatment to stimulate or restore the
ability of the immune system to fight infection and disease. Also
used to lessen side effects that may be caused by some cancer
treatments. Also known as immunotherapy, biotherapy, or biological
response modifier (BRM) therapy.
cancer: A term for diseases in which abnormal cells divide
without control. Cancer cells can invade nearby tissues and can
spread through the bloodstream and lymphatic system to other parts of
the body.
cancer vaccine: A form of biological therapy, which may
encourage a person's immune system to recognize cancer cells. These
vaccines may help the body reject tumors and prevent cancer from
recurring.
chemoprevention: The use of drugs, vitamins, or other
agents to try to reduce the risk of, or delay the development or
recurrence of, cancer.
chemotherapy: Treatment with anticancer drugs.
clinical trial: A research study that tests how well new
medical treatments or other interventions work in people. Each study
is designed to test new methods of screening, prevention, diagnosis,
or treatment of a disease.
combination chemotherapy: Treatment using more than one
anticancer drug.
combination therapy: The use of two or more modes of
treatment - surgery, radiotherapy, chemotherapy, immunotherapy - in
combination or alternately to achieve optimum results against
cancer.
Common Toxicity Criteria: A Web-based, interactive
application that uses standardized language to identify and grade
adverse events in cancer clinical trials.
NCI-designated Cancer Centers: There are 3 kinds of
NCI-designated cancer centers:
1. Comprehensive cancer centers, which conduct basic, clinical, and
preventive research programs, as well as community outreach and
education programs
2. Clinical cancer centers, which conduct primarily clinical research
programs but may have programs in other research areas as well
3. Cancer centers (formerly called Basic Science Cancer Centers),
which conduct basic or preventive research programs and do not have
clinical programs confidence intervals: These reflect a range of
values surrounding the true score that would be obtained if everyone
with a particular cancer were treated with the treatment under study.
The wider the interval, the more variable the result and the less
likely it is to be close to the true score. Confidence intervals are
typically thought of as the approximate bounds or limits surrounding
the true score. Researchers frequently use either a 95 or a 99
percent confidence interval.
control group: In a clinical trial, the group that receives
the accepted standard treatment being studied. In cases where no
standard treatment yet exists for a particular condition, the control
group would receive no treatment. No patient is placed in a control
group without treatment if there is any beneficial treatment known
for that patient. This group is compared to the group that receives
the investigational treatment. See also investigational group.
cooperative groups: Networks of institutions that jointly
carry out large clinical trials following the same protocols.
data and safety monitoring board (DSMB): An independent
committee whose membership includes, at minimum, a statistician and a
clinical expert in the area being studied. Members may also include
bioethicists or other clinicians knowledgeable about the trial's
subject matter. The National Institutes of Health requires DSMB
review of all phase 3 clinical trials. A DSMB might also review phase
1 or 2 trials that are blinded, take place at multiple locations, or
employ particularly high-risk interventions or vulnerable
populations.
diagnostic trial: A research study that evaluates methods
of detecting disease.
disease-free survival: The amount of time a participant survives
without cancer occurring or recurring, usually measured in
months.
double-blinded: A clinical trial in which neither the
medical staff nor the person knows which of several possible
therapies the person is receiving.
eligibility criteria: Participant eligibility criteria for
clinical trials can range from general (age, sex, type of cancer) to
specific (prior treatment, tumor characteristics, blood cell counts,
organ function). Eligibility criteria may also vary with trial phase.
In phase 1 and 2 trials, the criteria often focus on making sure that
people who might be harmed because of abnormal organ function or
other factors are not put at risk. Phase 2 and 3 trials often add
criteria regarding disease type and stage, and number of prior
treatments.
endpoint: What researchers measure to evaluate the results
of a new treatment being tested in a clinical trial. Research teams
establish the endpoints of a trial before it begins. Examples of
endpoints include toxicity, tumor response, survival time, and
quality of life.
Food and Drug Administration (FDA): A consumer protection
agency of the U.S. Department of Health and Human Services, FDA is
required by law to review all test results for new drugs to ensure
that they are safe and effective for specific uses.
gene: The functional and physical unit of heredity passed
from parent to offspring. Genes are pieces of DNA, and most genes
contain the information for making a specific protein.
gene therapy: Treatment that alters a gene. In studies of
gene therapy for cancer, researchers are trying to improve the body's
natural ability to fight the disease or to make the cancer cells more
sensitive to other kinds of therapy.
genetic: Inherited; having to do with information that is
passed from parents to offspring through genes in sperm and egg
cells.
genetic epidemiologic research: Research that involves
looking at tissue or blood samples from large populations of people
in order to determine how one's genetic make-up can influence
detection, diagnosis, prognosis, and ultimately, treatment.
genetics trials: Clinical trials that examine whether gene
transfer therapy can be used to treat cancer, or whether genetic
epidemiology research improves the understanding of cancer at the
cellular level. Actual genetic intervention (such as gene transfer)
trials are few in number, however trials are underway where actual
cellular manipulation at the gene level occurs.
imaging: Tests that produce pictures of areas inside the
body.
immunotherapy: See biological therapy.
informed consent: The process of providing all relevant
information about the trial's purpose, risks, benefits, alternatives,
and procedures to a potential participant, who then, consistent with
his or her own interests and circumstances, makes an informed
decision about whether to participate.
institutional review board (IRB): A board designed to
oversee the research process in order to protect participant safety.
Made up of researchers, ethicists, and laypeople from the community,
the board must review the trial protocols and the informed consent
forms participants sign.
intervention: The study agent or method that is being tested in a clincial trial or clinical study. The intervention is usually given to the investigational group while the control group receives standard treatment.
investigational group: In a clinical trial, the group that
receives the new agent being tested. See also control group.
investigational new drug (IND): A drug that the Food and
Drug Administration (FDA) allows to be used in clinical trials but
that the FDA has not approved for commercial marketing.
metastasis: The spread of cancer from one part of the body
to another. In cancer, metastasis is the migration of cancer cells
from the original tumor site through the blood and lymph vessels to
produce cancers in other tissues. Tumors formed from cells that have
spread are called "secondary tumors" and contain cells that are like
those in the original (primary) tumor. The plural is metastases.
metastatic cancer: Cancer that has spread from the place in
which it started to other parts of the body.
monoclonal antibodies: Laboratory-produced substances that
can locate and bind to cancer cells wherever they are in the body.
Many monoclonal antibodies are used in cancer detection or therapy;
each one recognizes a different protein on certain cancer cells.
Monoclonal antibodies can be used alone, or they can be used to
deliver drugs, toxins, or radioactive material directly to a
tumor.
multimodality therapy: Therapy that combines more than one
method of treatment.
National Cancer Institute (NCI): Part of the National
Institutes of Health of the United States Department of Health and
Human Services, is the Federal Government's principal agency for
cancer research. NCI conducts, coordinates, and funds cancer
research, training, health information dissemination, and other
programs with respect to the cause, diagnosis, prevention, and
treatment of cancer. Access the NCI Web site at www.cancer.gov.
New Drug Application (NDA): The application filed with FDA
by the trial sponsor once a trial has generated adequate data to
support a certain indication for a drug.
Office for Human Research Protections (OHRP): Safeguards
participants in federally funded research and provides unity and
leadership for 17 Federal departments and agencies that carry out
research involving human participants. OHRP enforces an important
regulation called the Common Rule, which sets standards for the
informed consent process; formation and function of IRBs; involvement
of prisoners, children, and other vulnerable groups in research; and
many other protective measures.
oncologist: A doctor who specializes in treating cancer.
Some oncologists specialize in a particular type of cancer treatment.
For example, a radiation oncologist specializes in treating cancer
with radiation.
p-value: A statistics term. A measure of probability that a
difference between groups during an experiment happened by chance.
For example, a p-value of .01 (p = .01) means there is a 1 in 100
chance the result occurred by chance. The smaller the p-value, the
more likely it is that the difference between groups was caused by a
difference between the tested treatments.
peer review: Scientific review by a panel of experts. The
primary responsibility of these experts is to assess the scientific
and technical merit of research proposals.
pharmacokinetics: The activity of drugs in the body over a
period of time, including the processes by which drugs are absorbed,
distributed in the body, localized in the tissues, and excreted.
phase 1 trial: Small groups of people with cancer are
treated with a certain dose of a new agent that has already been
extensively studied in the laboratory. During the trial, the dose is
usually increased group by group in order to find the highest dose
that does not cause harmful side effects. This process determines a
safe and appropriate dose to use in a phase 2 trial.
phase 2 trial: Phase 2 trials continue to test the safety
of the new agent and begin to evaluate how well it works against a
specific type of cancer. In these trials, the new agent is given to
groups of people with one type of cancer or related cancers, using
the dosage found to be safe in phase 1 trials.
phase 3 trial: Phase 3 studies are designed to answer
research questions across the disease continuum. Phase 3 trials
usually have hundreds to thousands of participants, in order to find
out if there are true differences in the effectiveness of the
treatment being tested.
phase 4 trial: Phase 4 trials are used to evaluate the
long-term safety and effectiveness of a treatment. Less common than
phase 1, 2, and 3 trials, phase 4 trials take place after the new
treatment has been approved for standard use.
Physician Data Query (PDQ): PDQ is an online database
developed and maintained by the National Cancer Institute. Designed
to make the most current, credible, and accurate cancer information
available to health professionals and the public, PDQ contains
peer-reviewed summaries on cancer treatment, screening, prevention,
genetics, and supportive care; a registry of cancer clinical trials
from around the world; and directories of physicians, professionals
who provide genetics services, and organizations that provide cancer
care.
placebo: An inactive substance that looks the same as, and
is administered in the same way as, a drug in a clinical trial. A
placebo may be compared with a new drug when no one knows if any drug
or treatment will be effective.
preclinical testing: A process in which scientists test
promising new cancer treatments in the laboratory and in animal
models. This is done to find out whether agents have an anticancer
effect and are safely tolerated in animals. Once an agent proves
promising in the lab, the sponsor applies for Food and Drug
Administration approval to test it in clinical trials involving
people.
prevention trials: Trials involving healthy people
who are at high risk for developing cancer. These trials try to
answer specific questions about and evaluate the effectiveness of
ways to reduce the risk of cancer.
principal investigator: The person responsible for
overseeing all aspects of a clinical trial, specifically, for
developing the concept and writing the protocol; submitting the
protocol for institutional review board approval; recruiting
participants; obtaining informed consent; and collecting, analyzing,
interpreting, and presenting data.
protocol: A written, detailed action plan for a clinical
trial. The protocol provides the background, specifies the
objectives, and describes the design and organization of the trial.
Every center participating in the trial uses the same protocol,
ensuring consistency of procedures and enhancing communication among
everyone working on the trial. This uniformity ensures that
participant information from all centers can be combined and
compared.
quality of life: The overall enjoyment of
life. Many clinical trials measure aspects of an individual's sense
of well-being and ability to perform various tasks to assess the
effects of cancer and its treatment on the overall quality of
life.
radiation therapy: The use of high-energy radiation
from x-rays, gamma rays, neutrons, and other sources to kill cancer
cells and shrink tumors. Radiation may come from a machine outside
the body (external-beam radiation therapy), or it may come from
radioactive material placed in the body in the area near cancer cells
(internal radiation therapy, implant radiation, or brachytherapy).
Systemic radiation therapy uses a radioactive substance, such as a
radiolabeled monoclonal antibody, that circulates throughout the
body. Also called radiotherapy.
randomization: A method used to prevent bias in research. A
computer or a table of random numbers generates treatment
assignments, and participants have an equal chance to be assigned to
one of two or more groups (e.g., the control group or the
investigational group).
randomized clinical trial: A study in which
the participants are assigned by chance to separate groups that
compare different treatments; neither the researchers nor the
participants can choose which group. Using chance to assign people to
groups means that the groups will be similar and that the treatments
they receive can be compared objectively. At the time of the trial,
it is not known which treatment is best. It is the patient's choice
to be in a randomized trial.
recurrence: The return of cancer, at the same site as the
original (primary) tumor or in another location, after the tumor had
disappeared.
regimen: A treatment plan that specifies the dosage, the
schedule, and the duration of treatment.
regression: A decrease in the size of a tumor, or in the
extent of cancer in the body.
relative risk: In cancer treatment trials, the
likelihood that cancer will recur within a specific timeframe in one
intervention group versus another.
remission: A decrease in or disappearance of signs and
symptoms of cancer. In partial remission, some, but not all, signs
and symptoms of cancer have disappeared. In complete remission, all
signs and symptoms of cancer have disappeared, although there still
may be cancer in the body.
risk/benefit ratio: The relation between the
risks and benefits of a given treatment or procedure. An
institutional review board, usually located where the clinical trial
is to take place, determines whether the risks in the trial are
reasonable with respect to the potential benefits. It is up to
individual potential participants to decide whether it is reasonable
for them in particular to participate.
sample size: In clinical trials, the number of
people participating in a trial.
screening trials: Clinical trials that assess the
effectiveness of new means of detecting cancer early in healthy
people, especially the earliest stages of cancer. For many types of
cancer, early detection results in improved outcomes. In addition,
these trials examine whether early treatment, as a result of early
detection, actually improves overall survival or disease-free
survival.
side effects: Problems that occur when treatment
affects healthy cells. Common side effects of cancer treatment are
fatigue, nausea, vomiting, decreased blood cell counts, hair loss,
and mouth sores.
single-blinded: Describes clinical trials set up in
such a way that participants do not know which therapy or
intervention they are receiving.
stage: The extent of a cancer, especially whether the
disease has spread from the original site to other parts of the body.
Numbers with or without letters are used to define cancer stages
(e.g., stage IIb).
standard treatment: A currently accepted and widely
used treatment for a certain type of cancer, based on the results of
past research.
statistical power: The chance of getting a
statistically significant result when there is one. Ideally, in
clinical trials statistical power should be .80 or .90--in other
words, there is an 80 to 90 percent chance that the true difference
in effectiveness between the treatments is the smallest size
considered medically important to detect.
statistically significant: Describes a mathematical
measure of difference between groups. The difference is said to be
statistically significant if it is greater than what might be
expected to happen by chance alone.
stratification: A separation process used in randomized
trials when factors that can influence the intervention's success are
known. For example, participants whose cancer has spread from the
original tumor site can be separated, or stratified, from those whose
cancer has not spread. Assignment of interventions within the two
groups is then randomized. Stratification enables researchers to look
in separate subgroups to see whether differences exist.
toxicity: Harmful side effects from an agent being
tested.
treatment group: See investigational group and
control group.
treatment trials: Treatment trials are designed to
test the safety and effectiveness of new drugs, biological agents,
techniques, or other interventions in people who have been diagnosed
with cancer. These trials evaluate the novel treatment against
standard treatment, if there is one.
tumor: An abnormal mass of tissue that results from
excessive cell division. Tumors perform no useful body function. They
may be benign (not cancerous) or malignant (cancerous).
vaccine: A substance or group of substances meant to cause
the immune system to respond to a cancer or to microorganisms, such
as bacteria or viruses.
Bibliography
American Cancer Society. (2001). Cancer facts and figures.
Atlanta, GA.
Bennett, C.L., Stinson, T. J., Vogel, V., Robertson, L., Leedy,
D., O'Brien, P., Hobbs, J., Sutton, T., Ruckdeschel, J. C., Chirikos,
T. N., Weiner, R. S., Ramsey, M. M., & Wicha, M. S. (2000).
Evaluating the financial impact of clinical trials in oncology:
Results from a pilot study from the Association of American Cancer
Institutes/Northwestern University Clinical Trials Costs and Charges
Project. Journal of Clinical Oncology, 18, 2805-2810.
Fireman, B., Fehrenbacher, L., Gruskin, E. P., & Ray, G. T.
(2000). Cost of care for patients in cancer clinical trials.
Journal of the National Cancer Institute, 92, 136-142.
Food and Drug Administration. (2001). www.fda.gov
[Web site].
Harris Interactive. (2001). Health Care News, 1(3).
[Poll]. Vol_iss3.pdf
National Cancer Institute. (2001). http://www.cancer.gov
[Web site].
National Cancer Institute. (2001). http://www.cancer.gov/search/cancer_literature/
[Web site].
National Cancer Institute. (2001). http://www.cancer.gov/clinicaltrials/
[Web site].
National Cancer Institute. (2001). http://ctep.info.nih.gov
[Web site].
Klimaszewski, A., Aikin, J., Bacon, M., DiStasio, S., Ehrenberger,
H., & Ford, B. (Eds.). (2000). Manual for clinical trials
nursing. Pittsburgh, PA: Oncology Nursing Press.
Shalala, D. (2000). Protecting research subjects - What must be
done. New England Journal of Medicine, 343, 808-810.
Wagner, J. L., Alberts, S. R., Sloan, J. A., Cha, S., Killian, J.,
O'Connell, M. J., Van Grevenhof, P., Lindman, J., & Chute, C. G.
(1999). Incremental costs of enrolling cancer patients in clinical
trials: A population-based study. Journal of the National Cancer
Institute, 91, 847-853.
|