
The information collection provisions in this guidance have been approved under OMB control number 0910-0577. This approval expires 1/31/2009. See additional PRA statement in Section IX of this guidance.
For questions regarding this document contact Casper Uldriks at the Center for Devices and Radiological Health (CDRH) at 240-276-0106, or at casper.uldriks@fda.hhs.gov.
 |
U.S. Department of Health and Human Services |
Written comments and suggestions may be submitted at any time for Agency consideration to the Division of Dockets Management, Food and Drug Administration, 5630 Fishers Lane, Room 1061, (HFA-305), Rockville, MD, 20852. When submitting comments, please refer to the exact title of this guidance document. Comments may not be acted upon by the Agency until the document is next revised or updated.
Additional copies are available from the Internet at: http://www.fda.gov/cdrh/comp/guidance/1217.pdf or to receive this document by fax, call the CDRH Facts-On-Demand system at 800-899-0381 or 301-827-0111 from a touch-tone telephone. Press 1 to enter the system. At the second voice prompt, press 1 to order a document. Enter the document number 1217 followed by the pound sign (#). Follow the remaining voice prompts to complete your request.
| This guidance represents the Food and Drug Administration's (FDA's) current thinking on this topic. It does not create or confer any rights for or on any person and does not operate to bind FDA or the public. You can use an alternative approach if the approach satisfies the requirements of the applicable statutes and regulations. If you want to discuss an alternative approach, contact the FDA staff responsible for implementing this guidance. If you cannot identify the appropriate FDA staff, call the appropriate number listed on the title page of this guidance. |
On October 26, 2002, section 301 of the Medical Device User Fee and Modernization Act of 2002 (MDUFMA) (Public Law 107-250) amended section 502 of the Federal Food, Drug, and Cosmetic Act (the Act) to require a device, or an attachment to the device, to bear prominently and conspicuously the name of the manufacturer, a generally recognized abbreviation of such name, or a unique and generally recognized symbol identifying the manufacturer. An important revision was made to section 502(u) of the Act by the Medical Device User Fee Stabilization Act of 2005 (MDUFSA) (Public Law 109-43), which became law on August 1, 2005.
MDUFSA amended section 502(u) by limiting the provision to reprocessed single-use devices (SUDs) and the manufacturers who reprocess them. Section 502(u) no longer sets forth requirements for original equipment manufacturers (OEMs), unless those manufacturers also reprocess single-use devices. Under the amended provision, if the original device or an attachment to it does not prominently and conspicuously bear the name of the manufacturer of the original device, a generally recognized abbreviation of such name, or a unique and generally recognized symbol identifying the manufacturer, the manufacturer who reprocesses the SUD may identify itself using a detachable label on the device’s packaging. The detachable label is intended to be affixed to the medical record of a patient by the user of the reprocessed SUD.
MDUFSA also requires that FDA issue guidance identifying the circumstances in which the name, abbreviation, or symbol of the manufacturer of an original device is not “prominent and conspicuous” under section 502(u) of the Act. On October 11, 2005, FDA issued draft guidance describing these circumstances. In addition, because section 502(u) requires that a reprocessed SUD or its attachment prominently and conspicuously bear the name of the reprocessor, except as described above, the document also provided guidance for reprocessors to determine whether their names, abbreviations, or symbols placed on the reprocessed SUDs are prominent and conspicuous.
MDFSA requires that FDA issue this guidance not later than 180 days after the date of enactment ( August, 1, 2005). Therefore, the agency requested that interested persons submit their comments on the draft guidance within 30 days of its issuance. As discussed below, FDA received comments on the draft, all of which were considered in finalizing the guidance.
FDA's guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidance documents describe the Agency's current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidance documents means that something is suggested or recommended, but not required.
FDA received comments from stakeholders, all of which were considered in developing this guidance. Each stakeholder who responded provided comments on the effective date for implementing the reprocessor labeling requirement. MDUFSA identifies two effective dates for compliance with section 502(u) of the Act.
The first effective date is August 1, 2006, which is 12 months after the date of enactment on August 1, 2005. This date applies to those reprocessed SUDs for which the OEM first marked the original device in a prominent and conspicuous manner before August 1, 2006. This date also applies to devices that are not marked or do not include an attachment with the OEM's name, as well as to devices which are marked or do include an attachment, but do not prominently and conspicuously bear the name of the OEM. Under section 502(u), the reprocessor may use a detachable label on such devices. Therefore, for all devices that are reprocessed and introduced or delivered for introduction into interstate commerce after August 1, 2006, the reprocessor must either mark the device, place its mark on an attachment, or where applicable, it may instead place its mark on a detachable label.
The second effective date relates to compliance in those situations where the OEM first marks its device after August 1, 2006. Two comments questioned this second effective date. The draft guidance provided that once the OEM marks the device, the reprocessor must mark the device, use an attachment, or where applicable, a detachable label. The comments argued that if the OEM marked its device after August 1, 2006, the reprocessor of the SUD should still have 12 months from the date on which the OEM first marked the device in which to mark the reprocessed device.
The statutory language in MDUFSA regarding the effective dates states that section 502(u) shall be effective with respect to reprocessed SUDs "12 months after the date of enactment of the Medical Device User Fee Stabilization Act of 2005, or the date on which the original device first bears the name of the manufacturer of the original device, a generally recognized abbreviation of such name, or a unique and generally recognized symbol identifying such manufacturer, whichever is later." (emphasis added). The statutory language does not provide for an additional 12 month period for the reprocessor to mark the device where the OEM first marks its product after August 1, 2006. The statutory language is clear that when an OEM prominently and conspicuously marks its device for the first time after August 1, 2006, the reprocessor -- who must have already identified itself through a detachable label as of August 1, 2006 if it did not mark the device or use an attachment -- does not get an additional 12 months in which to put its mark on the device itself.
The statutory language that requires the reprocessor's mark on "the date on which" the OEM first marks the device will give health care providers necessary information so they can report device related adverse events accurately and promptly to FDA. Mistakes in reporting and failures to make reports due to the inability to identify the correct manufacturer of a reprocessed single-use device undermine the agency’s postmarket surveillance program. Moreover, FDA believes that procedures for implementing compliant labeling specifications, among other manufacturing requirements, should already be established and implemented under a reprocessor’s Quality Systems (QS) program, as required by 21 CFR Part 820.
In addition to the above comments on the effective date, one comment questioned whether the statute requires that the detachable label be placed in the patient record. The statutory language clearly indicates that a detachable label is intended to be affixed to the medical record of the patient under section 502(u)(2), as amended.
One commenter requested that FDA include guidance on whether the mark of the manufacturer of the original device should be required to be obliterated by the reprocessor when placing its mark on the SUD. This comment also requested an exemption from device marking be permitted for comparative study purposes and that such studies not be considered to pose a significant risk under the Investigational Device Exemptions regulation (21 CFR 812). These issues fall outside the scope of this guidance and, therefore, are not addressed in this guidance.
The agency carefully considered all of the above comments. As discussed above, however, the information published in the draft guidance remains unchanged in this final guidance.For the purposes of this guidance, FDA has defined the following terms:
Attachment: An article secured to a device in such a way that it cannot be removed inadvertently.
Detachable label: A removable label on the device packaging that identifies the manufacturer who reprocessed the SUD and is intended to be affixed to the patient record.
Mark: A name, generally recognized abbreviation of such name, or a unique and generally recognized symbol that identifies a particular manufacturer.
Prominent and conspicuous: A manner of marking a device, as required by section 502(u) of the Act, such that the manufacturer’s mark is apparent to the user under ordinary conditions of use.
Reprocessor: A manufacturer who subjects a previously used SUD to additional processing and manufacturing for the purpose of an additional single use on a patient.
Single-Use Device: A device that is intended for one use, or on a single patient during a single procedure.This guidance applies to all manufacturers who reprocess single-use devices; therefore, it also applies to OEMs who reprocess SUDs.
The requirement that a reprocessed SUD, or an attachment to the SUD, must bear the reprocessor’s mark is effective on one of the following dates, whichever is later:
E.g., if the original device or an attachment to it bears the OEM’s mark prominently and conspicuously on July 1, 2006, then the reprocessed SUD or its attachment must prominently and conspicuously bear the mark of the reprocessor no later than August 1, 2006.
OR
For example, if the original device first prominently and conspicuously bears the OEM's mark on September 1, 2006, at that point in time a reprocessor must prominently and conspicuously use its own mark on the reprocessed device or its attachment before marketing.
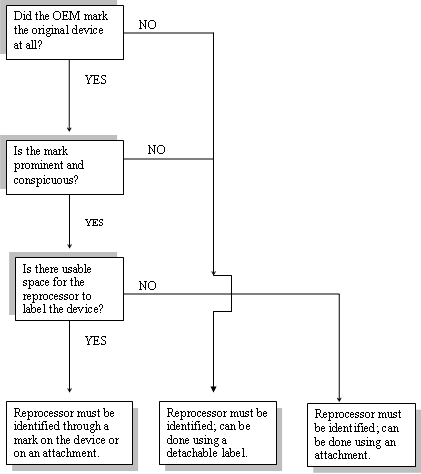
After August 1, 2006, even if the original device or an attachment to it does not bear the OEM’s mark (the OEM’s mark is absent or is not prominent and conspicuous), the reprocessed SUD must identify the reprocessor. Under this circumstance, the reprocessor may identify itself through use of a detachable label on the packaging of the SUD, as described below.
According to section 502(u) of the Act, a reprocessed SUD, or an attachment to it, must prominently and conspicuously bear the name of the manufacturer of the reprocessed device, a generally recognized abbreviation of the name, or a unique and generally recognized symbol identifying such manufacturer. The only exception to this requirement is when the original device, or an attachment to it, does not prominently and conspicuously identify the name of the original equipment manufacturer, a generally recognized abbreviation of the name, or a unique and generally recognized symbol identifying such manufacturer. Under this circumstance, the reprocessor may use a detachable label on the packaging to identify the manufacturer of the reprocessed device.
As stated in MDUFSA, the detachable label is intended to be affixed to the medical record of a patient. FDA therefore recommends that this label contain a statement directing a practitioner to remove the detachable label and affix it to the patient’s medical record when the reprocessed SUD is used.
If the original equipment manufacturer has marked the device in such a way that there is little or no usable space for a reprocessor to prominently and conspicuously mark the device, the reprocessor may satisfy the labeling requirement of section 502(u) through the use of an attachment to the device.
The following flow chart should help you decide whether you should place your mark on the device, use a detachable label, or use an attachment.
REPROCESSOR’S DECISION FLOW CHART
DO I PLACE MY MARK ON THE DEVICE, USE A DETACHABLE LABEL, OR USE AN ATTACHMENT?*
*Section 502(u) of the Federal Food, Drug, and Cosmetic Act, as amended.
No. Section 502(u) does not provide for a waiver from the labeling requirement.
This guidance contains information collection provisions that are subject to review by the Office of Management and Budget (OMB) under the Paperwork Reduction Act of 1995 (44 U.S.C. 3501-3520).
The time required to complete this information collection is estimated to average 1.0 hours per response, including the time to review instructions, search existing data resources, gather the data needed, and complete and review the information collection. Send comments regarding this burden estimate or suggestions for reducing this burden to:
Office of Compliance
Attention: Casper Uldriks
Center for Devices and Radiological Health, HFZ-300
Food and Drug Administration
2098 Gaither Road
Rockville , MD 20850
| An agency may not conduct or sponsor, and a person is not required to respond to, a collection of information unless it displays a currently valid OMB control number. The OMB control number for this information collection is 0910-0577, expires 01/31/2009. |
Updated May 1, 2006
![]()
CDRH Home Page | CDRH A-Z Index | Contact CDRH | Accessibility | Disclaimer
FDA Home Page | Search FDA Site | FDA A-Z Index | Contact FDA | HHS Home Page
Center for Devices and Radiological Health / CDRH



