SL770499
(ALFUZOSIN)
Date:
Alfuzosin is a selective a1–adrenergic blocker developed by
Sanofi-Synthelabo Inc. for the treatment of benign prostatic hyperplasia
(BPH). It was first approved for this
indication in
The benefits of alfuzosin treatment in symptomatic BPH include primarily: effective symptomatic relief of BPH; convenient once-daily (od) dosing; low incidence of class adverse reactions (e.g., dizziness and postural hypotension); treatment initiation at the therapeutic dose (i.e., no dose titration required); and no risk for sexual disturbances (e.g., impotence, libido decrease) observed with hormone-related treatment.
A phase III program using the 10 mg od regimen of alfuzosin was performed and a New Drug Application (NDA) was submitted to the FDA in December 2000 for the treatment of the signs and symptoms of BPH.
In October 2001, the FDA issued an “Approvable” letter indicating only 2 outstanding issues: 1) How to interpret results of the PKD4532 study, which assessed the effect of alfuzosin on QT interval using the Holter method and 2) The need to assess the possible interaction of alfuzosin with maximum doses of ketoconazole. No other issue was raised in the Approvable letter.
The FDA has focused this Advisory Committee meeting on the first issue. Therefore, this document focuses on the development program performed to assess the effect of alfuzosin on cardiac repolarization, specifically the evaluation of QT interval prolongation and methods used in this evaluation. The safety of alfuzosin relevant to cardiac QT interval prolongation is also documented.
Alfuzosin belongs to a drug class not suspected to cause ventricular arrhythmias. As an a1‑adrenergic blocker, alfuzosin is expected to increase heart rate (HR) in a dose‑dependent manner. This HR increase confounds assessment of a possible drug effect on QT interval, because QT interval is highly dependent on HR. The question, then, is how to correct for increased HR when assessing a possible effect on QT interval. The most widely used correction formulae (Bazett's [QTcB] and Fridericia's [QTcF]) attempt to normalize QT intervals to the values they would have if HR were 60 beats/min (bpm). These formulae over‑correct the QT interval when HR increases. Population and subject‑specific correction methods (QTcN and QTcNi, respectively) ([1],[2]) more precisely reflect the HR‑dependency of ventricular repolarization. Although not optimal, these methods more accurately predict the effect on QT interval of drugs that increase HR.
In studies assessing the effect of alfuzosin on QT interval that were submitted in the NDA (PKD4532 and PDY5105), the Holter monitoring method, specifically the Holter Bin method, was used. The Holter monitoring method, a recognized method for the assessment of QT interval ([3],[4]), was reported to be a valuable method in the recent preliminary concept paper for the clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non‑arrhythmic drugs ([5]). This method permits a direct comparison of QT intervals over a large range of HRs without need for correction. For this reason, Holter monitoring was used in these trials with alfuzosin.
For the alfuzosin dossier, 2 studies using both the Holter monitoring method and standard 12‑lead electrocardiograms (ECGs) were performed to thoroughly assess a possible effect of alfuzosin on QT interval. In both studies (PKD4532 and PDY5105), the Holter Bin method was applied to 12-lead ECG data and QTcB, QTcF, QTcN, QTcNi corrections were applied to ECG data. The first study (PKD4532) was a 4-way crossover study assessing 3 doses of alfuzosin (10 mg — i.e., the proposed therapeutic dose, 20 mg, and 40 mg) versus placebo in 24 healthy male volunteers. In this study, a 2 msec increase in QT interval according to the Holter Bin method was observed, whatever the dose of alfuzosin.
The second study (PDY5105) was subsequently designed after discussion and concurrence with the FDA. This study included a positive control, that induced modest increase in QT interval (>5‑10 msec) in order to validate both the study and the Holter Bin method. It was a 4-way crossover study conducted in 48 healthy volunteers. It assessed 2 doses of alfuzosin (10 mg and 40 mg) and a positive control, moxifloxacin (400 mg, the therapeutic dose) versus placebo. The 40 mg dose of alfuzosin was chosen to evaluate a possible effect on QT interval at a dose that covers the maximum plasma exposure anticipated to be encountered in clinical practice in patients with BPH, even when alfuzosin is co‑administered with ketoconazole. Moxifloxacin was selected as the positive control because it is known to induce a modest prolongation of the QT interval ([6]), but, to our knowledge, not to be associated with ventricular arrhythmias.
The design of PDY5105 was in agreement with the methodological recommendations underlined in the recent preliminary concept paper for the clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non‑arrhythmic drugs (5). In particular, in addition to inclusion of a positive control (moxifloxacin), a 1‑day assessment of QT interval under placebo allowed a time-matching of the comparative periods for drugs and placebo. The main criterion for evaluation in this study was QT interval assessed with the Holter Bin method (variation versus time‑matched baseline in comparison with placebo) over a 4‑hour period during which maximum plasma concentrations for both drugs were effectively achieved. The main results of this study are as follows:
· Moxifloxacin 400 mg produced a clear and statistically significant (p<0.0001) increase in QT/QTc interval, regardless of the assessment method used. This increase was in the range of those previously reported (6). The mean increase in QT interval compared to placebo was +7 msec with the Holter Bin method at a HR of 60 bpm.
· Alfuzosin 10 mg (the therapeutic dose) did not induce significant changes in QT interval, regardless of the assessment method used. The mean change in QT interval compared to placebo was +0.1 msec with the Holter Bin method at a HR of 60 bpm.
· Alfuzosin 40 mg (4 times the therapeutic dose) induced a weak, albeit statistically significant, increase in QT interval compared to placebo (mean equal to +2.9 msec) with the Holter Bin method at a HR of 60 bpm. This increase was half of that observed with moxifloxacin at its therapeutic dose.
· As expected, alfuzosin increased HR in a dose-dependent manner. In addition, moxifloxacin also slightly increased HR. The HR increase with alfuzosin 40 mg explains the over-correction observed with QTcB and QTcF. A similar observation can be made for alfuzosin 10 mg and moxifloxacin, although the differences are less pronounced. QTcN and QTcNi approaches showed values intermediate between those recorded with the QTcB and QTcF and the Holter method.
These results are consistent with those from the PKD4532
study. The minor differences in results,
which are within the range of the sensitivity of the method, are attributed to
an improved study design in the second study.
From the results of these 2 studies, it is concluded that:
· The Holter Bin method, which allows comparison of direct QT interval measurements, is reproducible, sensitive, capable of demonstrating a statistically significant change in QT interval as small as 2 to 3 msec, and is appropriate to assess drugs that modify HR.
· The £2.9 msec mean increase in QT interval recorded with alfuzosin at 4 times the therapeutic dose is less than that observed with moxifloxacin at its therapeutic dose. This small increase in QT interval is below the range suspected to be associated with torsades de pointes [preliminary concept paper (5)].
These conclusions are corroborated by the large clinical experience on the safety profile of alfuzosin in patients with BPH, a population of male patients, generally older than 65 years-of-age and suffering in more than 40% of cases from associated cardiovascular diseases. Specifically:
· No signal regarding cardiac arrhythmia related to QT/QTc interval prolongation with alfuzosin was detected in the clinical trials safety database (more than 2000 patients treated with alfuzosin in phase II/III controlled trials).
· A strong confirmation of this statement is provided by the large post‑marketing experience with alfuzosin (more than 130,000 patients in cohort observational surveys and a number of days of therapy superior to 1350 millions). No arrhythmogenic signal is detected in a population unrestricted for prescription.
In conclusion:
· The Holter Bin method is confirmed as a valuable method to assess the effect on QT interval of a drug that affects HR.
· Thorough clinical assessment of the effect of alfuzosin on QT interval is not indicative of a prolongation of QT interval that could be associated with ventricular arrhythmia. This conclusion is corroborated by the absence of a signal of ventricular arrhythmia in a large clinical and post‑marketing experience.
Title page.................................................................................................................................. 1
Executive summary.................................................................................................................... 2
Table of contents....................................................................................................................... 5
List of tables.............................................................................................................................. 7
List of figures............................................................................................................................ 8
List of abbreviations.................................................................................................................. 9
1. introduction...................................................................................................... 10
2. preclinical and pharmacokinetic characteristics................. 12
2.1 Preclinical data (HERG channel)................................................................................ 12
2.2 Relevant pharmacokinetic data.................................................................................... 12
3. methods for qt evaluation...................................................................... 15
3.1 Bazett and Fridericia correction formulae................................................................... 15
3.2 Population and subject specific formulae.................................................................... 16
3.3 Holter-monitoring method............................................................................................ 18
3.3.1 Data acquisition........................................................................................................ 18
3.3.2 Data processing........................................................................................................ 18
3.3.3 Holter Bin method validation.................................................................................... 20
3.4 Use of a positive control.............................................................................................. 21
4. Intensive ECG monitoring studies (pkd4532 and pdy5105)........... 21
4.1 Study design (PDY5105)............................................................................................. 21
4.1.1 Rationale for the choice of the alfuzosin 40 mg
dose................................................ 22
4.1.2 Primary method for QT assessment (Holter Bin method).......................................... 23
4.1.3 Secondary method for QT assessment (12-lead ECG).............................................. 24
4.1.4 Sample size calculation and statistical method......................................................... 24
4.2 Results......................................................................................................................... 25
4.2.1 Baseline HR and QT values...................................................................................... 26
4.2.2 Heart rate results....................................................................................................... 27
4.2.3 QT/QTc interval results............................................................................................ 27
4.2.3.1 Moxifloxacin.......................................................................................................... 28
4.2.3.2 Alfuzosin................................................................................................................ 28
4.2.3.3 PKD4532 QT results............................................................................................. 29
4.2.4 Outliers..................................................................................................................... 30
4.2.5 PK/PD relationship................................................................................................... 31
4.3 Conclusion................................................................................................................... 32
5. clinical sAFETY.................................................................................................. 32
5.1.1 Methodology............................................................................................................. 32
5.1.2 Clinical trials............................................................................................................ 33
5.1.3 Post-marketing experience........................................................................................ 33
5.1.3.1 Cohort observational surveys................................................................................ 34
5.1.3.2 Spontaneous reporting............................................................................................ 34
5.1.3.3 Post-marketing experience conclusion................................................................... 35
5.1.4 Overall clinical safety conclusion............................................................................ 35
6. Overall interpretation............................................................................. 35
6.1 Validity of the approach............................................................................................... 35
6.1.1 Approaches to the correction of QT interval for drugs
that affect HR...................... 35
6.1.2 Clinical trial design for the assessment of QT interval
prolongation........................ 36
6.2 The risk of cardiac arrhythmia associated with these
results....................................... 37
7. Supportive information.............................................................................. 38
7.1 Summaries of study reports.......................................................................................... 38
7.1.1 Summary of INT5056................................................................................................ 38
7.1.2 Summary of PKD4532.............................................................................................. 41
7.1.3 Summary of PDY5105.............................................................................................. 45
7.2 Preclinical safety......................................................................................................... 51
7.3 Study design................................................................................................................. 51
7.4 QT interval assessment................................................................................................ 53
7.5 Clinical
safety.............................................................................................................. 57
8. references........................................................................................................... 58
Table (4.2.1) 1
- Baseline HR and QT interval values (mean ± SEM) for each treatment group
(Study PDY5105).................................................................................................... 26
Table (4.2.2) 2
- 12-lead ECG: HR change
from baseline for alfuzosin 10 and 40 mg and moxifloxacin 400 mg versus
placebo, pairwise comparisons (Study PDY5105).............. 27
Table (4.2.3.1) 1
- QT interval change from baseline for moxifloxacin 400 mg compared
to placebo (Study PDY5105)................................................................................................. 28
Table (4.2.3.2) 1
- QT interval change from baseline for alfuzosin 10 mg compared to
placebo (Study PDY5105)................................................................................................. 29
Table (4.2.3.2) 2
- QT interval change from baseline for alfuzosin 40 mg compared to
placebo (Study PDY5105)................................................................................................. 29
Table (4.2.3.3) 1
- QT interval change for alfuzosin 10 mg, 20 mg, and 40 mg
compared to placebo (Study PKD4532)................................................................................................. 30
Table (7.2) 1
- Inhibition of HERG potassium current expressed in CHO cells................ 51
Table (7.3) 1
- Study details of PDY5105 and PKD4532.................................................. 52
Table (7.4) 1
- Holter‑monitoring method – 4 hour-period: QT interval change from baseline of
alfuzosin 10 mg, alfuzosin 40 mg, and moxifloxacin 400 mg, in
comparison with placebo - primary endpoints (Study PDY5105)......................................................... 53
Table (7.4) 2 - 12‑lead ECG: change from baseline to Cmax: moxifloxacin 400 mg versus
placebo, pairwise comparisons (Study PDY5105)............................................................ 53
Table (7.4) 3 - 12‑lead ECG: change from baseline to Cmax: alfuzosin 10 mg and 40 mg versus
placebo, pairwise comparisons (Study PDY5105)................................................ 54
Table (7.4) 4 - Individual data of subjects with
QTcB>450 msec and/or delta QTcB>60 msec (Study PDY5105)..................................................................................................... 56
Table (7.5) 1 - Number (%) of patients experiencing
AEs that are potentially suggestive of a proarrhythmic effect in double‑blind
controlled phase II/III studies in BPH patients – alfuzosin all
formulations................................................................................................... 57
Table (7.5) 2
- Number of SAEs that are potentially suggestive of a proarrhythmic effect
collected in cohort observational surveys or spontaneously reported with
alfuzosin, displayed by MedDRA preferred terms............................................................................. 57
Figure (2.2) 2
- Representation of increases in alfuzosin Cmax and AUC related
to various intrinsic and extrinsic factors after administration of 10 mg
alfuzosin............................... 14
Figure (3.1) 1
- 12-lead ECG: relationship
between QTcB and QTcF change from baseline and RR change from baseline on
placebo data (Study PKD4532)....................... 16
Figure (3.2) 1
- 12-lead ECG: relationship between QTcN (study population-specific)
and QTcNi (subject-specific) change from baseline and RR change from baseline
on placebo data (Study PKD4532)....................................................................................................... 17
Figure (3.3.2) 1
- Holter‑monitoring analysis of ECG complexes..................................... 19
Figure (4.1.1) 1
- Alfuzosin Cmax and AUC values after alfuzosin 10 mg
tablet according to intrinsic and extrinsic factors............................................................................................. 23
Figure (4.2) 1
- Mean alfuzosin and moxifloxacin plasma concentrations versus time
profiles observed after a single administration of alfuzosin 10 mg,
alfuzosin 40 mg, or moxifloxacin 400 mg (Study PDY5105)............................................................................ 26
Figure (4.2.5) 1
- Relationship between QTcNi change from baseline and mean plasma
concentration using data from alfuzosin 10 mg and 40 mg and placebo
(Study PDY5105) 31
Figure (7.4) 1 - 12-lead ECG: relationship between QTcB and QTcF change from
baseline and RR change from baseline on placebo data (Study PDY5105)....................... 54
Figure (7.4) 2 - 12-lead ECG: relationship between QTcN and QTcNi change
from baseline and RR change from baseline on placebo data (Study PDY5105)....................... 55
|
5ARIs |
5a-reductase inhibitors |
|
AE |
adverse event |
|
ANOVA |
analysis of variance |
|
AUC |
area under the plasma concentration versus time curve extrapolated to infinity |
|
AUClast |
area under the plasma concentration versus time curve calculated using the trapezoidal method from time zero to the real time tlast |
|
bid |
twice daily |
|
BMI |
body mass index |
|
BPH |
benign prostatic hyperplasia |
|
CHO |
Chinese hamster ovary |
|
CI |
confidence interval |
|
Cmax |
maximum plasma concentration |
|
Ctrough |
plasma concentration observed before treatment administration during repeated dosing |
|
CYP3A4 |
cytochrome P450 3A4 |
|
DBP |
diastolic blood pressure |
|
ECG |
electrocardiogram |
|
FDA |
Food and Drug Administration |
|
HERG |
human ether-a-go-go-related gene |
|
HPLC |
high performance liquid chromatography |
|
HR |
heart rate |
|
IC20 |
concentration inducing a 20% inhibition |
|
IC50 |
half maximal inhibitory concentration |
|
ISHNE |
International Society for Holter and Noninvasive Electrocardiology |
|
LC-MS/MS |
liquid chromatography tandem mass spectrometry |
|
LUTS |
lower urinary tract symptoms |
|
NDA |
new drug application |
|
od |
once daily |
|
PCSA |
potentially clinically significant abnormality |
|
SAE |
serious adverse event |
|
SBP |
systolic blood pressure |
|
SD |
standard deviation |
|
SEM |
standard error of the mean |
|
TEAE |
treatment emergent adverse event |
|
tid |
three times a day |
|
t1/2z |
elimination half-life |
|
tmax |
first time to reach Cmax |
|
ULN |
upper limit of normal |
|
US |
United States |
|
UV |
ultraviolet |
1. 000introduction
Alfuzosin hydrochloride is a selective a1‑adrenergic blocker developed by Sanofi‑Synthelabo Inc. for the treatment of BPH.
Lower urinary tract symptoms (LUTS), resulting from bladder outlet obstruction associated with BPH, are common in older men and have a significant impact on their daily lives ([7]). Medical treatment is the first-line option for symptomatic and uncomplicated BPH. The 2 principal options for the medical management of BPH are a1‑adrenergic blockers and 5a‑reductase inhibitors (5ARIs). 5ARIs block the conversion of testosterone to dihydrotestosterone, resulting in a reduction in prostate volume. a1‑adrenergic blockers act on the dynamic component of obstruction by decreasing the sympathetically controlled tone of prostatic smooth muscle. The latter are characterized by a rapid onset of action, with evidence of efficacy from the first dose. Thus, a1‑adrenergic blockers have a first‑line role in the pharmacotherapeutic management of BPH patients who are symptomatic but who are not candidates for surgery.
Alfuzosin was first approved for use in Europe for the BPH indication in 1987 at a dose of 2.5 mg three times a day (tid). Subsequently, 5 mg twice daily (bid) and 10 mg once daily (od) regimens were developed and approved. Alfuzosin is now approved in 108 countries worldwide, including all of Europe, Canada, and Australia. It has provided over 1350 million therapy days of treatment for BPH from the first marketing through December 2002. Alfuzosin has never been marketed for hypertension.
The therapeutic value of alfuzosin for treatment of LUTS in BPH has been well established ([8],[9],[10],[11],[12],[13]). In addition, its therapeutic interest in the management of acute urine retention related to BPH has also been reported ([14]). The benefits of alfuzosin treatment in symptomatic BPH include primarily: effective symptomatic relief of BPH; convenient od regimen; low incidence of class adverse reactions (e.g., dizziness and postural hypotension); dose initiation with the therapeutic dose (i.e., no titration required); no risk for the sexual disturbance (impotence, libido decrease) observed with hormone‑related treatment.
There are 3 other a1‑adrenergic blockers approved for BPH treatment in the United States (US): HytrinÒ (terazosin) for BPH and hypertension was first approved in 1987 ([15]); CarduraÒ (doxazosin) for BPH and hypertension was first approved in 1990 ([16]); and FlomaxÒ (tamsulosin) for BPH was first approved in 1997 ([17]). NDA data concerning the effect of these drugs on cardiac repolarization is not known to the authors of this document.
In 1996, Sanofi-Synthelabo Inc. began a separate stand-alone phase III clinical development program for the od regimen to support an NDA in the US. While the phase III program was in progress, it became apparent to the company that a high‑dose ECG study, to demonstrate the lack of effect of drug on cardiac repolarization, was becoming a requirement for all drugs submitted for NDAs in the US. An earlier clinical pharmacology ECG study, however, suggested that the effect of the HR increase produced by alfuzosin on the calculation of the QT interval length (using the Bazett or Fridericia traditional universal formulae) was confounding the ability to clearly assess the potential of alfuzosin to affect cardiac repolarization. To avoid this confounding influence of HR, a method based on Holter monitoring was chosen to assess the effect of alfuzosin on QT interval. The Holter Bin method, used in the trial, permits the direct comparison of treatments at the same HR, avoiding the need for arbitrary correction formulae.
An NDA for the use of 10 mg od alfuzosin was submitted to the FDA in December 2000 for the treatment of the signs and symptoms of BPH. The NDA provided the results of the ECG study (PKD4532) that used the Holter Bin method to assess effects on QT interval‑length prolongation. This study compared placebo to the therapeutic dose (10 mg) and to supra-therapeutic doses (20 mg and 40 mg). The FDA declared this NDA “Approvable” in October 2001 and, in a subsequent meeting in January 2002, clarified the following issues that were presented in the Approvable letter:
· The interpretation of the results of the PKD4532 study that assessed the effect of alfuzosin on QT interval using the Holter Bin method.
· The assessment of the possible interaction of alfuzosin with maximum doses of ketoconazole.
No other issue was raised in the Approvable letter.
Both issues were subsequently addressed by developing a complementary clinical program that comprised 2 studies (INT5056 and PDY5105). These studies were designed and conducted with FDA concurrence to address the outstanding concerns.
· A new interaction study used the maximum permitted dose of ketoconazole (INT5056).
· A new high-dose ECG study (PDY5105) compared placebo to the therapeutic (10 mg) and the supra-therapeutic (40 mg) doses of alfuzosin and to the US‑approved therapeutic dose (400 mg) of moxifloxacin as a positive control. Moxifloxacin is known to modestly increase QT interval length when assessed using correction formulae. This study was similar in design to the previous ECG study (PKD4532), except that the assessment of moxifloxacin by 12-lead ECG and the Holter Bin method allowed comparison of the signal from both the Holter method and correction formulae for moxifloxacin, bridging the 2 methods. The study design complied with the FDA requirement — i.e., adequately powered with positive control and placebo capable of demonstrating small QT effects (at least 5 msec). The study design received FDA concurrence in May 2002.
Related to the assessment of alfuzosin on QT interval, the FDA has convened the Advisory Committee meeting 29 May 2002 to discuss: 1) clinical trial designs for assessment of QT prolongation, 2) approaches to the correction of QT interval for drugs that affect HR, and 3) risks of cardiac arrhythmia associated with different degrees of QT prolongation.
Therefore, this document focuses on the development program performed to assess the effect of alfuzosin on cardiac repolarization, specifically the evaluation of QT interval prolongation and the methods used for this evaluation. The safety of alfuzosin relevant to cardiac QT prolongation is also documented.
2. 000preclinical and pharmacokinetic characteristics
2.1 000Preclinical data (HERG channel)
The effects of alfuzosin and other reference drugs that belong to the same therapeutic class were evaluated on human ether-a-go-go-related gene (HERG) potassium channels, which are stably expressed in Chinese hamster ovary (CHO) cells.
Alfuzosin has a negligible inhibitory effect on the
HERG potassium current, with an IC50=83.3±16.6 µmol/L [Table (7.2) 1]. The first observed inhibition (IC20)
occurred at 10 µmol/L. In human,
alfuzosin is 90% bound to plasma protein, so the free fraction is around
1.4 ng/mL at maximum plasma concentration (Cmax) (i.e.,
3.6 nmol/L). This concentration is
2,800 times lower than the IC20.
2.2 000Relevant pharmacokinetic data
Figure (2.2) 1
displays the plasma concentration versus time profile for alfuzosin
10 mg after single dose administration.

Figure (2.2) 1 - Mean (SD) alfuzosin plasma concentration-time profile obtained in healthy male volunteers after a single administration of 10 mg alfuzosin
Alfuzosin is slowly absorbed for up to 20 hours. Peak plasma concentrations reach a plateau from 5 to 12 hours after administration. The elimination half-life of alfuzosin after the administration of a 10 mg tablet is approximately 10 hours. There is only a small accumulation of exposure as a result of repeated administration (approximately a 1.1‑fold increase) and steady state is observed after the second day of administration.
Alfuzosin is eliminated primarily by metabolism, with only 11% of the administered dose excreted in the urine as unchanged compound. Cytochrome P450 3A4 (CYP3A4) is the principle hepatic enzyme involved in the metabolism of alfuzosin. The elimination half‑life of total radioactivity after 14C-alfuzosin administration is similar to the elimination half-life of alfuzosin itself. This indicates that metabolites should not accumulate after repeated administration. The metabolites are rapidly excreted, resulting in alfuzosin being the major circulating compound in the plasma. Alfuzosin and its metabolites are excreted in feces (69% of dose) and urine (24% of dose).
The FDA Approvable letter (05 October 2001) requested a study
to assess the possible interaction between the therapeutic dose of alfuzosin
(10 mg) and the highest recommended dose of ketoconazole (400 mg).
The results of this study (INT5056) indicate that repeated
administration of 400 mg ketoconazole once daily over 8 days under
fed conditions resulted in a 2.3‑ and 3.0‑fold increase in
alfuzosin Cmax and area under the plasma concentration versus
time curve extrapolated to infinity (AUC),
respectively (see study summary in Section 7.1.1).
Figure (2.2) 2 presents intrinsic (repeat administration, age, renal impairment, hepatic impairment) and extrinsic (inhibition of its metabolism) factors that increase exposure of alfuzosin (see Section 4.1.1).

* (estimated from Ctrough values in BPH patients)
** (estimated from changes in clearance)
Figure (2.2) 2 - Representation of increases in alfuzosin Cmax and AUC related to various intrinsic and extrinsic factors after administration of 10 mg alfuzosin
3. 000methods for qt evaluation
3.1 000Bazett and Fridericia correction formulae
Alfuzosin and all other a1‑adrenergic blocker drugs are expected to increase HR. The HR increase observed after administration of an a1‑adrenergic blocker is the consequence of a primary vasodilatory effect followed by a neural sympathetic stimulation. The magnitude of this baroreflex response is subject to large inter-individual variability. Increases in mean HR, as well as large inter-individual HR variability, were in fact observed following alfuzosin administration during the 2 completed phase I ECG studies.
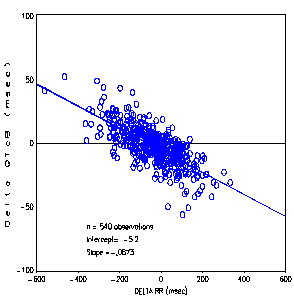
Because the QT interval is HR-dependent, an evaluation of drug-induced QT interval changes must always accommodate for the underlying changes in HR. Many general HR correction formulae have been proposed to normalize the QT interval to a HR of 60 bpm, using several mathematical functions to correct for HR changes [please see review in (1,[18])]. The value of these “universal” (i.e., the same correction is applied to every individual) correction formulae has been discredited ([19]), because they substantially over‑correct or under-correct the QT interval when HR moves away from normal resting ranges ([20],[21]). This is particularly true for the most widely used correction formula proposed by Bazett (QTcB=QT/RR0.5) and, to a lesser extent, for the Fridericia formula (QTcF=QT/RR0.33). With the Bazett correction formula, QT interval is clearly under‑corrected when HR is under 60 bpm and over-corrected when HR is higher than 60 bpm ([22]). Moreover, this over-correction increases as HR moves away from 60 bpm ([23]). Using the placebo data from the PKD4532 study, the problems associated with the Bazett and Fridericia formulae when HR changes are illustrated in Figure (3.1) 1. This figure shows that the corrected QT interval remains strongly HR dependent, because the regression line of the changes in QTc interval as a function of changes in HR would have been expected to be horizontal for an adequate correction. A similar relationship using placebo data was also shown for the PDY5105 study, as illustrated in Figure (7.4) 1.
QTcB QTcF

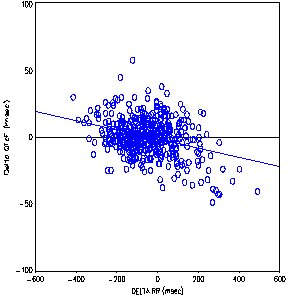
Figure (3.1) 1 - 12-lead ECG: relationship between QTcB and QTcF change from baseline and RR change from baseline on placebo data (Study PKD4532)
Therefore, the Bazett's and Fridericia's correction formulae confound efforts to evaluate an increase in QT interval in the presence of HR increases.
3.2 000Population and subject specific formulae
Malik and co-authors proposed a study population-specific correction formula to better correct for HR variation for the population studied (1,2). This population‑specific correction formula (QTcN) is based on a simple parabolic model in which the exponent is estimated from the model fit to the population. Baseline or placebo QT/RR data are used to produce the exponent a, leading to a correction formula of generic form QTcN=QT/RRa (1). This study population-specific QT correction formula was recently proposed to the FDA during 2 Advisory Committee meetings [ziprazidone ([24]) and telithromycin ([25])].
The study population-specific QT interval correction formula (QTcN) does not adequately fit all subjects (1,2,18,[26]), so the concept of a subject-specific correction formula (QTcNi) was subsequently proposed (26) in order to minimize the correction error. This method is based on several ECG recordings at different HRs in the same individual that are used to determine the specific QT/RR relationship for that individual.
Population- and subject-specific correction formulae
were generated and used in the 2 thorough ECG studies (PKD4532 and
PDY5105) using multiple, resting, 12‑lead ECGs recorded during the
placebo period.
In the subject-specific approach, data were fitted to a parabolic model that yielded individual exponential factors ranging from 0.188 to 0.278. These formulae allow a better correction of QT intervals for HR changes [Figure (3.2) 1] compared with the Bazett or Fridericia formulae [Figure (3.1) 1]. In some subjects, however, the observed QT/RR values deviate from the parabolic fit, in agreement with the findings of Malik and co‑authors (26). Thus, although performing better than previous formulae, the subject‑specific correction (Ni) still does not fully account for changes in HR. A similar relationship using placebo data was also shown for the PDY5105 study, as illustrated in Figure (7.4) 2.
QTcN QTcNi


Figure (3.2) 1 - 12-lead ECG: relationship between QTcN (study population-specific) and QTcNi (subject-specific) change from baseline and RR change from baseline on placebo data (Study PKD4532)
3.3 000Holter-monitoring method
As none of the correction formulae provide optimal ways for QT interval assessment for drugs that increase HR, other approaches based on continuous Holter monitoring have been developed. Continuous ECG recordings allow:
· To build individualized QT/RR relationships for each subject,
· To perform, within each subject, a direct comparison of the uncorrected QT interval, between baseline placebo and drug treatment at identical HR.
This latter approach, hereafter referred to as the Holter Bin method, does not require a correction over a large range of HRs and was therefore chosen as the primary method used in the thorough ECG studies with alfuzosin (PKD4532 and PDY5105).
The Holter Bin method of analysis of QT interval complies with the recommendations of the International Society for Holter and Noninvasive Electrocardiology (ISHNE) Task Force ([27]) and has been previously used to assess QT intervals in clinical pharmacology studies (3,4,[28]).
A description of the data acquisition, data processing, and method validation for the Holter Bin method is provided below.
3.3.1 000Data acquisition
Continuous Holter recordings were obtained during a placebo run-in day and an under‑treatment day. The procedures used to evaluate each subject were as follows:
· Continuous ECGs (over approximately 24 hours) were recorded using a 3‑lead Holter digital device (Syneflash Digital Recorder, Ela Medical),
· Electrodes were positioned at the same place on the chest wall (positions were identified and marked),
· Digital storage of the information was obtained on computer CD-ROM that was transferred to an expert cardiologist.
3.3.2 000Data processing
The expert cardiologist performed all readings in a blinded manner through the use of validated software (WinAtrec®). Per period, about 98% of the recorded complexes were readable and were retained for classification and averaging. Test protocols with acceptance conditions were applied to validate the various steps of data processing and analysis (Section 3.3.3).
Data processing was performed in 3 steps:
1) RR interval measurements,
2) Classification of all sinus cardiac ECG complexes into 10 msec RR groups (“bins”),
3) Averaging of complexes within each RR bin and measurement of QT intervals.
The QT interval length of the averaged complex was the value for each subject in each treatment group (or baseline) used in the analysis.
A schematic diagram of the data processing is presented in Figure (3.3.2) 1.

Figure (3.3.2) 1 - Holter‑monitoring analysis of ECG complexes
The result of the blinded data processing was the average QT interval for each RR bin, and each treatment period, by subject.
3.3.3 000Holter Bin method validation
The systems used in the implementation of the Holter Bin monitoring method have been the subject of extensive validation. The goal was to ensure that all computer systems used in the process meet state-of-the-art international requirements, in terms of specifications, operational qualification, documentation, traceability, security, and audit trails. The objective of the validation plan was to ensure proper functionality and accuracy of methodology embedded in the application. To achieve this objective, a dedicated simulated ECG was designed to replicate most important conditions.
The data acquisition systems used in the study (Syneflash Digital recorder, ELA Medical) and standard 12-lead ECGs (MAC 5000, Marquette) were validated systems compliant with international norms and Good Clinical Practice standards, and were approved by the FDA. The key software equipment used for the method was WinAtrec v4.0 (AMPS LLC). It was intended that this software provide a tool to build templates for QT interval analyses from the Holter recordings.
Specifically, the following critical points were qualified:
· Verification of data access authorization at all stages of use,
· Presence of audit trails,
· Verification of production of the data bins,
· Verification of detection of abnormal ECGs,
· Verification of calculations and associated documentation.
Validation of the algorithm used to obtain averaged ECG templates has been previously described (3). Only at the end of the qualification phases and after examination of the full and satisfactory validation dossier was the form documenting final acceptance of the system signed‑off. Beyond system validation, several other requirements were necessary for a complete validation of the method, including the determination of accuracy, precision, limit of detection, specificity, reproducibility, and robustness. From previously published studies using this approach (4,[29]) and from the first alfuzosin ECG placebo‑controlled study (PKD4532), the precision and robustness of the method were demonstrated. In order to demonstrate accuracy, specificity, and reproducibility, however, a carefully designed placebo‑controlled study with inclusion of a positive control was needed. This was one of the goals of study PDY5105.
3.4 000Use of a positive control
The preliminary concept paper (5) encourages use of a concurrent positive control group to evaluate the effect of a drug on QT interval and to support the use of novel correction approaches, like the Holter-based correction. Use of a positive control demonstrates the ability of the experiment to detect relevant effects on QT/QTc intervals (i.e., validates the study design, assesses the sensitivity of the approach used, and avoids false negatives). Moreover the positive control chosen should be one that consistently produces a mean QT/QTc interval effect in the range of 5-10 msec, without inducing a large increase in HR. Another important requirement for proper use of the positive control is to conduct the study in a double-blind manner, and to ensure that the QT intervals are measured in a blinded manner by an expert cardiologist.
Moxifloxacin was a good positive
control candidate because it induces a consistent and modest increase in QT/QTc
intervals after administration of a single dose of 400 mg (6). Following FDA concurrence with the choice of
moxifloxacin as an adequate positive control, the specifications of the
criterion to be met to validate the Holter Bin method, as requested in the
October 2001 approvable letter, were agreed with FDA. The prospective
goal of detecting a statistically significant increase in QT intervals of at
least 5 msec in the moxifloxacin group was established as a basis of the
PDY5105 study design.
4. 000Intensive ECG monitoring studies (pkd4532 and pdy5105)
Two studies (PKD4532 and PDY5105) were performed to thoroughly assess the effect of alfuzosin on QT interval. A summary and study design details for these 2 studies are provided in Section 7.1.2, Section 7.1.3, and Table (7.3) 1, respectively. The first study, PKD4532, was a four‑way, double‑blind, crossover study assessing 3 single doses of alfuzosin (10 mg, 20 mg, and 40 mg) versus placebo in 24 healthy volunteers. The following sections will primarily focus on the second study (PDY5105).
4.1 000Study design (PDY5105)
The second study, PDY5105, was designed with the FDA’s concurrence. This study was a 4‑way crossover study assessing 2 single doses of alfuzosin (10 mg and 40 mg) and a single dose of the positive control (400 mg moxifloxacin) versus placebo in 48 healthy volunteers. The study was a single-center, randomized, double‑dummy and placebo‑controlled, and used standard 12‑lead ECG as well as Holter monitoring. Each period consisted of a 2-day run-in placebo followed by a single-dose day, with a washout of 5 to 9 days between successive periods. A thorough pharmacokinetic assessment was carried out with blood samples collected at the same time as ECG recordings. The 12‑lead ECG reading was performed in a blinded centralized manner by qualified cardiologists. The Holter monitoring was analyzed blindly and independently of the 12‑lead ECG reading, which was analyzed by another qualified cardiologist.
A significant difference between the 2 studies was the use in PDY5105 of a 2‑day placebo run-in period (Day 1 and Day 2) preceding the day of drug administration (Day 3), whereas in PKD4532 each period consisted of a single day of treatment. Thus in PDY5105, Holter recording performed at Day 2 was considered as the baseline for the Holter recording performed on the day of dosing. The only available baseline in study PKD4532 is the Holter recorded at the day of screening. This substantial improvement in PDY5105 study design allowed a more relevant and time-matched baseline, increasing the sensitivity of the trial.
4.1.1 000Rationale for the choice of the alfuzosin 40 mg dose
A high dose of 40 mg alfuzosin (i.e., 4 times the therapeutic dose) was utilized in PDY5105 as well as in the previous PKD4532 study. This dose gives exposure greater than that achieved at the therapeutic dose (10 mg) with any identified intrinsic (age, renal impairment, hepatic impairment) or extrinsic (inhibition of its metabolism) factors that increase the exposure of alfuzosin [Figure (4.1.1) 1]. In addition, at a dose of 40 mg, the mean Cmax and the upper 95% confidence interval (CI) for Cmax were equivalent to or greater than those predicted in BPH patients who are more than 75 years old or who have moderate renal insufficiency and who are also administered ketoconazole. Therefore, the 40 mg dose utilized in PDY5105 and PDY4532 evaluates alfuzosin exposure at the upper limits of what might be observed in the target population.
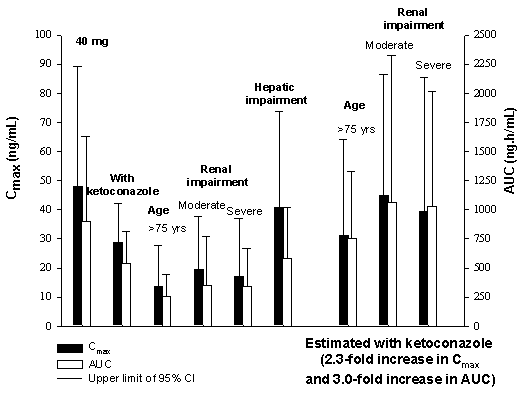
Figure (4.1.1) 1 - Alfuzosin Cmax and AUC values after alfuzosin 10 mg tablet according to intrinsic and extrinsic factors
4.1.2 Primary method for QT assessment (Holter Bin method)
Holter recordings for the primary analysis were made for moxifloxacin, alfuzosin, and placebo during a 4‑hour period when the highest drug concentrations were anticipated (i.e., T7 through T11 hours). The baseline was defined for each period as the time‑matched interval (T7-T11 h) of the Day 2 placebo run‑in.
Changes in QT intervals were calculated for each subject and for each RR bin between the treatment period and the baseline (placebo run-in) to generate the following primary endpoints:
· 1000 msec RR bin (QT1000) corresponding to 60 bpm HR,
· Largest sample size RR bin: for the RR bin containing the largest total number of complexes,
· Average of all RR bins.
4.1.3 Secondary method for QT assessment (12-lead ECG)
The conditions of recording and measurements were as follows:
Standard 12-lead ECGs were digitally recorded using a MAC 5000 (Marquette, USA) electrocardiographic machine after the subject had rested for at least 10 minutes. The ECGs (each consisting of a 10 sec recording) were performed at specific time points corresponding to the PK sampling times. A computer-assisted, manual, on-screen measurement of digitized ECG wave-forms was performed from the digital records, using a standard methodology: on either lead II (preferably) or V2 to V5 (same lead for a given subject) the QT interval was determined by the tangent method ([30]). RR (msec), PR (msec), QRS intervals (msec), and QT interval duration (msec) were measured on 3 consecutive complexes and averaged.
HR, QTcB, QTcF, QTcN and QTcNi were derived from the mean values of the measured parameters.
The analysis variable was change from baseline for HR, QT and QTcB, QTcF, QTcN, and QTcNi at the time of individual Cmax and at hours 7 through 11 (period corresponding to the same period of Holter monitoring analysis). The baseline value for these analyses was the mean of the 3 measures on Day 3-T0 of each period.
4.1.4 Sample size calculation and statistical method
Study PDY5105 was designed
to provide sufficient power to evaluate both primary (Holter Bin method) and
secondary (standard 12-lead ECG) pharmacodynamic endpoints. As indicated in the first study (PKD4532),
the statistical power to detect a given difference with the Holter Bin method
is much greater than that with the standard 12‑lead ECG. Therefore, the sample size calculation is
driven by the 12-lead ECG.
Estimates of SD for both endpoints were obtained from the previous study, PKD4532. For standard 12‑lead ECGs, it was determined that a minimum of 36 completed subjects would be required to provide 80% power to detect a 5 msec difference in QTc interval from baseline between active treatment and placebo, assuming a within-subject standard deviation of 7.5 msec (the SDwithin obtained for QTcN in PKD4532 was 7.8 msec). A sample size of 45 subjects would provide 80% power to detect a 7.5 msec difference from baseline between active treatment and placebo assuming a standard deviation of 12.5 msec (the SDwithin obtained for QTcB in PKD4532 was 12 msec).
For the Holter Bin method primary endpoint, a sample size of at least 36 subjects would provide 88% power to detect a 3 msec difference in QT interval between active treatment and placebo, assuming a 4 msec SDwithin for change from baseline in QT interval at RR=1000 ms (the SDwithin obtained for QT interval change from baseline at in PKD4532 was 3.6 msec).
A sample size of 45 subjects would provide 80% power to detect a 3 msec difference in QT interval between active treatment and placebo, assuming a 5 msec SDwithin for change from baseline in QT interval at RR =1000 ms (upper bound of the 95% CI for SDwithin).
The primary statistical method was prospectively defined as follows:
Mean QT interval change from baseline for each endpoint was analyzed by a mixed linear‑effects model including terms for subject as random effect, and sequence, period, and treatment as fixed effects. Pairwise comparisons were performed using linear contrasts as follows: moxifloxacin 400 mg versus placebo, alfuzosin 10 mg versus placebo, and alfuzosin 40 mg versus placebo.
Results from these comparisons are presented as the mean difference versus placebo with its 95% CI.
4.2 000Results
The primary focus of the results presented below is PDY5105, as this confirmatory study had an improved design, a larger sample size (48 subjects versus 24) and included a positive control. A summary table of the core results of PKD4532 study is also provided.
Forty-eight male subjects (mean age 27 years; range 19-45 years) were included in the study and analyzed for safety. Three subjects withdrew for personal reasons (not safety related) after receiving 1 or 2 doses of run‑in placebo. Forty‑five subjects received an active treatment, completed the study, and were evaluated for the pharmacodynamic and pharmacokinetic analyses.
Figure (4.2) 1 presents the PK profile for alfuzosin 10 mg and 40 mg and moxifloxacin 400 mg. It confirms that the Holter Bin method and 12-lead ECG analyses were performed at the time of Cmax for both drugs, as prospectively planned.

Figure (4.2) 1 - Mean alfuzosin and moxifloxacin plasma concentrations versus time profiles observed after a single administration of alfuzosin 10 mg, alfuzosin 40 mg, or moxifloxacin 400 mg (Study PDY5105)
4.2.1 000Baseline HR and QT values
Table (4.2.1) 1 shows that pre-dose baseline values of HR and QT were comparable across treatment groups.
Table (4.2.1) 1 - Baseline HR and QT interval values (mean ± SEM) for each treatment group (Study PDY5105)
|
Baseline |
Parameter |
Placebo |
Alfuzosin 10 mg |
Alfuzosin 40 mg |
Moxifloxacin 400 mg |
|
Holter Bin method Run-in placebo 7 to 11 hours |
HR (bpm) |
60.9 ± 1.2 |
60.4 ± 1.2 |
61.8 ± 1.2 |
61.6 ± 1.2 |
|
QT1000 (msec) |
398.9 ± 3.2 |
404.0 ± 3.5 |
403.5 ± 3.5 |
404.7 ± 3.1 |
|
|
12‑lead ECG data pre-dose (H0) |
HR (bpm) |
54.7 ± 1.4 |
54.8 ± 1.4 |
55.0 ± 1.2 |
54.3 ± 1.2 |
|
Uncorrected QT (msec) |
407.1 ± 4.3 |
407.4 ± 4.2 |
405.7 ± 4.1 |
408.9 ± 4.3 |
Baseline values should not be directly compared between methods (Holter Bin method and 12‑lead ECG) as they were evaluated at different periods of time.
4.2.2 Heart rate results
Table (4.2.2) 2 presents the differences versus placebo in HR changes from baseline for the 3 treatment groups, as assessed from 12-lead ECG, either during the [7-11h] time window or at time of individual Cmax.
Moxifloxacin 400 mg increased mean HR by 1.5 to 2.8 bpm versus placebo. Alfuzosin 10 and 40 mg increased HR by 1.5 to 5.2 bpm and 3.7 to 5.8 bpm, respectively, versus placebo.
Table (4.2.2) 2 - 12-lead ECG: HR change from baseline for alfuzosin 10 and 40 mg and moxifloxacin 400 mg versus placebo, pairwise comparisons (Study PDY5105)
|
Assessment
Period |
Treatment |
P-Value |
Mean Difference vs Placebo (bpm) |
95% CI |
|
|
Lower Bound |
Upper Bound |
||||
|
[H7, H8, H9,
H10, H11] |
Alfuzosin 10 mg |
0.0569 |
1.5 |
-0.0 |
3.0 |
|
Alfuzosin 40 mg |
0.0001 |
3.7 |
2.1 |
5.2 |
|
|
Moxifloxacin 400
mg |
0.0594 |
1.5 |
-0.1 |
3.0 |
|
|
At time of
individual Cmax |
Alfuzosin 10 mg |
0.0013 |
5.2 |
2.2 |
8.3 |
|
Alfuzosin 40 mg |
0.0001 |
5.8 |
3.2 |
8.4 |
|
|
Moxifloxacin 400
mg |
0.0005 |
2.8 |
1.3 |
4.2 |
|
Although, these mean changes versus placebo appear not to be very large, there is clearly a larger increase with alfuzosin 40 mg than with alfuzosin 10 mg. There is also a modest HR increase observed with moxifloxacin.
In addition, the proportion of subjects with an increase in HR (from baseline) over 10 bpm was 20%, 14%, 27%, and 47% for placebo, moxifloxacin, alfuzosin 10 mg and alfuzosin 40 mg, respectively. The proportion of subjects with an increase over 15 bpm was 33% in the alfuzosin 40 mg group, as compared to 9% in the placebo group.
These results indicate that, because alfuzosin induces a marked HR increase in a substantial number of subjects, there is a real potential for traditional correction formulae (especially QTcB and QTcF, to a lesser extent QTcN and QTcNi) to result in biased overestimation of QT changes.
4.2.3 QT/QTc interval results
For the Holter Bin method, the focus will be mainly on the results corresponding to the RR=1000 msec (QT1000 corresponding to a HR of 60 bpm), as the usual correction formulae (QTcB, QTcF, QTcN and QTcNi) also use the HR of 60 bpm as the reference for QT correction. The results for the 2 other Holter Bin endpoints are essentially similar and are presented in Table (7.4) 1.
Results are presented as QT changes from baseline for alfuzosin 10 mg, alfuzosin 40 mg, and moxifloxacin 400 mg, in comparison with placebo over the 7-11 hours time window.
For the standard 12‑lead ECGs, results from the 7 to
11 hour time window are presented below.
The results obtained at the time of Cmax for each individual
are presented in Table (7.4) 2
and Table (7.4) 3.
4.2.3.1 000Moxifloxacin
Moxifloxacin 400 mg (the therapeutic dose) produced a clear and highly statistically significant (p<0.0001) increase in QT interval, regardless of the method used [Table (4.2.3.1) 1]. These results are within the range of published data (6).
Moxifloxacin slightly increased mean HR versus placebo by 1.5 bpm, thereby explaining the over‑correction observed with the Bazett and Fridericia formulae.
Table (4.2.3.1) 1 - QT interval change from baseline for moxifloxacin 400 mg compared to placebo (Study PDY5105)
|
Method |
Endpoints |
Mean Difference Between Moxifloxacin 400 mg and
Placebo (msec) |
95% CI |
P value |
|
Holter Bin method 7 to 11 hours |
QT1000 |
+ 7.0 |
4.4 ; 9.6 |
0.0001 |
|
12-lead ECG [H7, H8, H9, H10,
H11] |
QTcNi |
+ 9.4 |
6.9 ; 11.8 |
0.0001 |
|
QTcN |
+ 9.4 |
6.9 ; 11.9 |
0.0001 |
|
|
QTcF |
+10.3 |
7.7 ; 13.0 |
0.0001 |
|
|
QTcB |
+11.9 |
8.3 ; 15.6 |
0.0001 |
4.2.3.2 000Alfuzosin
· Alfuzosin 10 mg (i.e., the therapeutic dose) did not induce any statistically significant change in the QT interval, regardless of the method used [Table (4.2.3.2) 1]. The mean HR increase of 1.5 bpm compared to placebo during the 7 to 11 hour time window explains the over‑correction observed with the Bazett and Fridericia formulae.
Table (4.2.3.2) 1 - QT interval change from baseline for alfuzosin 10 mg compared to placebo (Study PDY5105)
|
Method |
Endpoints |
Mean Difference Between Alfuzosin 10 mg and
Placebo (msec) |
95% CI |
P value |
|
Holter Bin method 7 to 11 hours |
QT1000 |
+ 0.1 |
-2.5 ; 2.6 |
0.97 |
|
12-lead ECG [H7, H8, H9, H10,
H11] |
QTcNi |
+ 0.5 |
-2.0 ; 2.9 |
0.70 |
|
QTcN |
+ 0.5 |
-2.0 ; 3.0 |
0.71 |
|
|
QTcF |
+ 1.6 |
-1.1 ; 4.3 |
0.24 |
|
|
QTcB |
+ 3.3 |
-0.3 ; 6.9 |
0.07 |
· Alfuzosin 40 mg (i.e., 4 times the therapeutic dose) induced a weak, albeit statistically significant, QT increase of 2.9 msec when assessed with the Holter Bin method [Table (4.2.3.2) 2]. However, this increase was half of that induced by moxifloxacin at its therapeutic dose.
The mean HR increase of 3.7 bpm compared to placebo during the 7 to 11 hour time window, which was associated with the substantial number of subjects (33%) experiencing an HR increase by more than 15 bpm, explains the large over‑correction observed with the Bazett and Fridericia formulae.
Table (4.2.3.2) 2 - QT interval change from baseline for alfuzosin 40 mg compared to placebo (Study PDY5105)
|
Method |
Endpoints |
Mean difference Between Alfuzosin 40 mg and
Placebo (msec) |
95% CI |
P value |
|
Holter Bin method 7 to 11 hours |
QT1000 |
+ 2.9 |
0.3 ; 5.5 |
0.03 |
|
12-lead ECG [H7, H8, H9, H10,
H11] |
QTcNi |
+ 4.7 |
2.2 ; 7.1 |
0.0003 |
|
QTcN |
+ 4.6 |
2.1 ; 7.0 |
0.0004 |
|
|
QTcF |
+ 6.9 |
4.2 ; 9.5 |
0.0001 |
|
|
QTcB |
+10.8 |
7.2 ; 14.4 |
0.0001 |
4.2.3.3 000PKD4532 QT results
Results from study PDY5105 are consistent with those obtained in the previous ECG study (PKD4532). In PKD4532, a 2 msec increase for QT1000 (Holter Bin method used for primary analysis) was observed, whatever the dose of alfuzosin (10, 20, and 40 mg) [Table (4.2.3.3) 1].
Table (4.2.3.3) 1 - QT interval change for alfuzosin 10 mg, 20 mg, and 40 mg compared to placebo (Study PKD4532)
|
Method |
Endpoints |
Treatment |
Mean Difference
(msec) |
95% CI |
P value |
|
Holter Bin method 7 to 11 hours |
QT1000 |
Alfuzosin 10 mg |
+ 2.0 |
- ; 3.2 |
0.0110 |
|
QT1000 |
Alfuzosin 20 mg |
+ 1.9 |
- ; 3.6 |
0.0664 |
|
|
QT1000 |
Alfuzosin 40 mg |
+ 1.7 |
- ; 3.2 |
0.0590 |
The slight numerical differences observed between the 2 studies are in the range of variability that can be seen between 2 studies, reaching also the limit of sensitivity of the method. They are also likely due to differences in design (no run-in placebo in PKD4532) leading to analyses with different baselines in the 2 studies. This improvement in the design of study PDY5105, associated with the use of a positive control and the larger sample size, resulted in increased precision for this study.
4.2.4 000Outliers
Outlier subjects were analyzed according to standard categorical analyses of QTc interval data (absolute values QTc>450 msec and changes from baseline QTcB>60 msec).
In total, 7 subjects had a value of QTcB over 450 msec at some point during the study (maximum value observed was 458 msec). These 7 subjects experienced this increased (in the range of 450-458 msec) QTcB while on placebo (2 subjects), moxifloxacin (1 subject), alfuzosin 10 mg (1 subject), and alfuzosin 40 mg (3 subjects).
Four subjects had delta QTcB intervals over 60 msec (3 on alfuzosin 40 mg and 1 on alfuzosin 10 mg).
In the alfuzosin-treated group, all subjects having either QTcB>450 msec or delta QTcB>60 msec had large HR increases versus baseline (17 to 49 bpm). For these subjects, at the corresponding RR bin (when available), the absolute QT values assessed by the Holter Bin method were not higher than 423 msec, and QT changes from baseline were no more than 5 msec [Table (7.4) 4].
No QTcF value over 450 msec or delta QTcF over 60 msec was recorded, whatever the treatment.
No QTcN or QTcNi value over 450 msec or a delta QTcF over 60 msec was recorded, whatever the treatment.
Furthermore, the arrhythmia analysis performed on the 24-hour Holter recording did not show the presence of any ventricular ectopic beat, including in the alfuzosin 40 mg group at the time of maximal plasma concentration.
4.2.5 000PK/PD relationship
PK/PD relationship for alfuzosin was explored by using data from 12-lead ECG measured at the time of blood sample collection. As QTcNi has been shown earlier to be the QT interval correction formula that best accounts for HR, it will be used as the pharmacodynamic variable.
All data points are used in this analysis, resulting in 1470 observations in the alfuzosin figure (including placebo data).
It is apparent from Figure (4.2.5) 1 that there is a large intrinsic variability of QTcNi, as shown by the placebo data (0 concentration). Furthermore, it is noteworthy that there is no increase of more than 35 msec in QTcNi. The relationship between QTcNi and alfuzosin plasma concentration appears very shallow. At the highest concentrations obtained in the study (above 80 ng/mL, i.e., the mean Cmax of alfuzosin 40 mg), there appears to be no QTcNi increase.

Figure (4.2.5) 1 - Relationship between QTcNi change from baseline and mean plasma concentration using data from alfuzosin 10 mg and 40 mg and placebo (Study PDY5105)
4.3 000Conclusion
From this confirmatory study (PDY5105), the following conclusions can be drawn:
· Using the 12‑lead ECG, moxifloxacin produced statistically significant increases of more than 9 msec in QTc interval (range from 9.4 to 11.9 msec), regardless of the correction formula. The values observed with Bazett’s and Fridericia’s formulae are comparable to values reported in the literature and validate the study.
· Using the Holter Bin method, moxifloxacin (400 mg) produced a statistically significant QT1000 increase of about 7 msec.
· Using the same method, the therapeutic dose of alfuzosin produced no significant change in the QT interval. At 4 times the therapeutic dose, alfuzosin produced a mean QT1000 change of 2.9 msec, less than half what is observed with moxifloxacin.
· This study indicates that the Holter Bin method can detect a statistically significant change in QT interval as small as 3 msec. This demonstrates that the Holter Bin method is sensitive and can detect moderate drug‑induced increases of QT interval.
5. 000clinical sAFETY
In controlled phase II/III clinical trials conducted in Europe and the US, 2104 patients suffering from BPH received alfuzosin: 1012 patients received alfuzosin od, 685 patients received alfuzosin 2.5 mg tid, and 407 patients received alfuzosin 5 mg bid. Post‑marketing experience including cohort observational surveys (more than 130,000 patients) and spontaneous reporting (nearly 1350 million therapy‑days of commercial use) since the launch in Europe in 1988 have provided a very substantial source of information for the assessment of a potential signal of an arrhythmogenic risk.
5.1.1 000Methodology
Adverse events (AEs) that are potentially suggestive of a proarrhythmic effect have been quoted in the preliminary concept paper “The clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs" dated 15 November 2002, revised 28 January and 06 February 2003”. The concept paper distinguishes AEs reported in clinical studies and in post‑marketing:
· In clinical trials, the rate of some rare AEs is to be compared to the control group (torsades de pointes, ventricular tachycardia, ventricular arrhythmia, ventricular ectopy, ventricular fibrillation and flutter, cardiac arrest, sudden death, syncope, dizziness, palpitations, and seizures).
· In post‑marketing, the available data should be examined for evidence of QT/QTc interval prolongation and torsades de pointes and for AEs possibly related to QT/QTc interval prolongation, such as cardiac arrest, sudden cardiac death, ventricular arrhythmias (e.g., ventricular tachycardia and ventricular fibrillation). A well‑characterized episode of torsades de pointes has a high probability of being related to drug use, whereas the other events that are reported more commonly may be of significance if seen in a population at low risk for them (e.g., young men experiencing sudden death).
Therefore, the safety database was reviewed accordingly:
· In controlled clinical studies, all AEs possibly related to QT/QTc interval prolongation were reviewed, whatever the causal relationship to study drug and whatever the seriousness criterion.
· In the post-marketing experience, serious adverse events (SAEs) reported to Sanofi‑Synthelabo Pharmacovigilance up to 31 December 2002 were reviewed, whatever the causal relationship.
5.1.2 000Clinical trials
The characteristics of the BPH population included in phase II/III controlled clinical studies were: men with a mean age of 64±7 (³65: 47%), 25% suffering from concomitant hypertension, 46% reporting having a coronary artery disease at baseline, and 48.5% of whom had a creatinine clearance at baseline below 80 mL/mn (12.5% below 60 mL/mn).
No cases of torsades de pointes, ventricular fibrillation, or ventricular tachycardia have been reported during the clinical development of alfuzosin, regardless of the formulation or dose used. The most frequently reported AEs potentially suggestive of a proarrhythmic effect, as identified in the preliminary concept paper, were dizziness (3.5%) and syncope (0.3%) [Table (7.5) 1]. These events are well known to be due to the vasodilatory effect of the a1‑adrenergic blocker class of drugs (15,16,17,[31]).
In other studies (phase I, extension studies, phase IV, and studies in non‑BPH indication), including 7084 volunteers or patients, no cases of torsades de pointes or ventricular fibrillation were reported. Two elderly volunteers experienced asymptomatic, non‑polymorphic, transient non-sustained ventricular tachycardia (14 and 17 beat run, 12 and 17 hours post 15 mg od dosing, respectively), a common ECG finding in elderly volunteers ([32]). The only fatal case (i.e., sudden death in a 73 year‑old patient receiving 5 mg bid) was reported in a phase IV non-placebo controlled study. He had been recently diagnosed with angina pectoris and left ventricular hypertrophy requiring nitrates.
5.1.3 000Post-marketing experience
As the first launch of alfuzosin occurred in 1988, an extensive post‑marketing experience was recorded from cohort observational surveys and from post‑marketing surveillance. Taking into account the treated population, i.e., men 65 years-of-age and over in 68% of the cases (European IMS data, Jan-Dec 2002), having co-morbidity in 68.5% of cases, including 40% underlying cardiovascular diseases, such as hypertension (23%) or coronary artery disease (8.5%), and taking concomitant drugs in 67% of cases ([33]), the recording of cardiovascular events was expected. The interpretation of these events should always take into consideration the medical context and the age of the treated population.
5.1.3.1 000Cohort observational surveys
More than 130,000 patients were exposed up to 3 years (mean duration 9 months) to different alfuzosin formulations in cohort observational surveys up to 31 December 2002. The surveys were not designed to specifically monitor safety. The number of SAEs involving ventricular rhythm disorders or which might be related to QT/QTc interval prolongation according to the preliminary concept paper and recorded by Sanofi-Synthelabo Pharmacovigilance are presented in Table (7.5) 2.
These data show:
· No cases of torsades de pointes, ventricular tachycardia, or QT/QTc interval prolongation were reported.
· One case of ventricular fibrillation (possibly related to a myocardial infarction) in a patient with risk factors for coronary heart disease was reported.
· Seventeen fatalities related to a cardiac disorder (cardiac arrest, sudden cardiac death) were reported in observational surveys. In 14 out of the 17 patients, a medical history of cardiovascular disorders was mentioned (coronary heart disease, myocardial infarction, hypertension, or cardiac failure).
· Twenty-three other reports of death, for which either no specific cause or circumstances were noted.
In conclusion, taking into account the elderly population treated with alfuzosin in general practice, we detected no signal of a potential arrhythmogenic risk.
5.1.3.2 000Spontaneous reporting
Spontaneously reported SAEs recorded in the Sanofi‑Synthelabo Pharmacovigilance database transmitted by either the health professional or health authorities or published in the literature from launch up to 31 December 2002 were reviewed. The estimated number of therapy days with alfuzosin (all formulations) up to 31 December 2002 is about 1350 million.
· Neither torsades de pointes nor QT interval prolongation was reported.
· Seven cases of ventricular arrhythmias (ventricular tachycardia: N=3, ventricular fibrillation: N=4) were reported. In 1 case, the role of alfuzosin was excluded (the AE occurred 16 days after the last intake of alfuzosin) and in the remaining 6 reports, all patients were suffering from pre-existing cardiac disorders.
· Cardiac arrests following the first intake of a 2.5 mg tablet of alfuzosin were reported in 2 patients (71 and 79 years-of-age) treated with concomitant antihypertensive and vasodilatory drugs. Both patients recovered.
· Five cases of sudden death in patients aged between 56 and 83 years were noted. In all cases except 1, either a family history of sudden death or medical history of cardiac disorder or myocardial infarction were reported by the notifiers.
Overall, the reporting rate of spontaneous cases of events potentially related to QT/QTc interval prolongation is estimated to be 1 per 100 million therapy days, and the rate of fatality (N=7) is estimated to be 0.6 per 100 million therapy days. This low rate clearly does not provide a signal for an arrhythmogenic potential of alfuzosin, especially in a middle-aged/elderly male population frequently presenting concomitant risk factors.
5.1.3.3 000Post-marketing experience conclusion
Post-marketing information has been gathered on alfuzosin since the first launch in 1988. Alfuzosin is marketed in 108 countries worldwide, with an experience of nearly 1350 million therapy days. From this vast experience, no arrhythmogenic signal has been detected:
· from cohort observational surveys (more than 130,000 patients),
· from spontaneous notification from health professionals,
· from reports from national health authorities, or
· from cases in the literature.
5.1.4 000Overall clinical safety conclusion
· No signal regarding cardiac arrhythmia related to QT/QTc interval prolongation with alfuzosin was detected in the clinical trials safety database.
· Furthermore, a strong confirmation of this statement is provided by the large post‑marketing experience with alfuzosin in a population unrestricted for prescription.
6. 000Overall interpretation
6.1 000Validity of the approach
The validity of our approach on the potential effects of alfuzosin on cardiac repolarization can be critically reviewed in light of the recent preliminary concept paper issued by the FDA on “The clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non‑antiarrhythmic drugs”.
6.1.1 000Approaches to the correction of QT interval for drugs that affect HR
In both studies evaluating the effects of alfuzosin on QT interval, the magnitude of HR changes is not compatible with the use of Bazett’s and Fridericia’s formulae, which over‑correct at increased HRs.
The inability of the Bazett’s and Fridericia’s correction factors to fully compensate for the effects of HR on the QT interval was demonstrated by the strong correlation between QT and RR interval established using the placebo data from PKD4532 and PDY5105.
The Holter Bin method, on which our primary analyses is based, allows a direct comparison between QT interval under treatment and QT interval under placebo at similar HR, thereby avoiding the use of a correction factor.
The QT variations obtained for both the positive control (moxifloxacin) and the 2 doses of alfuzosin using the Holter Bin method are in closer agreement with those obtained using population-specific (QTcN) and individual-specific (QTcNi) correction methods, which are other methods developed to accommodate for changes in HR.
6.1.2 000Clinical trial design for the assessment of QT interval prolongation
The design of study PDY5105 was developed with the help of experts and with concurrence from the FDA. The design of this study was in agreement with the methodology recommendations proposed in the recent concept paper (5).
Main features of the
study design
PDY5105 was a 4‑way crossover, single-dose, randomized, double-dummy, and placebo‑controlled study. The 4 treatments were placebo, alfuzosin at 10 and 40 mg, and moxifloxacin (400 mg). A 1-day assessment of QT interval under placebo allowed a time matching of the comparative periods for treatment and placebo. In addition, blood samples were collected at the time of ECG recordings.
Positive controls
Moxifloxacin was a good positive control candidate because it induces a consistent and modest increase in QT/QTc intervals after administration of a single dose of 400 mg. In PDY5105, moxifloxacin provided increases in QTcB and QTcF in line with previously published studies using standard ECG techniques.
Statistical power
This study was powered to detect QT interval changes of 3 msec and of 5 msec (Holter Bin method and standard resting ECG analysis, respectively).
Dose selection
In study PDY5105 a potential dose effect was investigated by administering alfuzosin doses of 10 mg and 40 mg.
The 40 mg dose (4 times the therapeutic dose) allowed the exploration of concentrations higher than those achieved following therapeutic dose administration. This dose produced plasma concentrations greater than any factor shown to increase exposure to alfuzosin at therapeutic doses. A single dose was chosen because there is little accumulation of alfuzosin after repeated administration.
The maximum dose of alfuzosin used was close to the maximum tolerated dose and was agreed upon with the FDA. The administration of doses higher than 40 mg could have led to major vasodilatory effects in some healthy volunteers.
The design of the first study (PKD4532) was essentially similar to PDY5105, although it did not include a positive control and a 1-day placebo run-in. According to current methodological standards, the design of these 2 studies is appropriate for the thorough assessment of QT interval variations.
6.2 000The risk of cardiac arrhythmia associated with these results
Data from the 2 studies discussed in this document consistently show that the effect on QT interval assessed using the Holter Bin method was in the range of 2 to 3 msec, even at doses up to 4 times the therapeutic dose of alfuzosin. Using the same Holter Bin method, moxifloxacin at the therapeutic dose of 400 mg increased QT interval by 7 msec.
It is recognized by the recent concept paper that drugs that produce a maximum effect on QT interval of less than 5 msec have not at this time been associated with torsades de pointes. This fact has recently been reported in a paper published by Shah ([34]).
Study PDY5105 demonstrates that the increase in QT interval for alfuzosin was less than for moxifloxacin, a drug already approved. Further, this 2 to 3 msec QT increase for alfuzosin was below the 5 msec threshold quoted in the concept paper as being without repolarization risk.
The results of the Holter Bin method were robust and in the same range as results using the subject-specific correction method (QTcNi).
The large safety experience with alfuzosin corroborates the lack of signal of arrhythrogenic risk seen in the ECG studies performed. No signal regarding cardiac arrhythmia related to QT/QTc interval prolongation with alfuzosin was detected in the clinical trials safety database. Furthermore, a strong confirmation of this statement is provided by the large post‑marketing experience with alfuzosin.
In conclusion, thorough clinical assessment of the effect of alfuzosin on QT interval is not indicative of a prolongation of QT interval that could be associated with ventricular arrhythmia. This conclusion is corroborated by the absence of a signal of ventricular arrhythmia in a large clinical and post‑marketing experience