Many gene–disease associations proposed to date have not been consistently replicated across different populations. Nonreplication often reflects false positives in the original claims. However, occasionally, nonreplication may be due to heterogeneity due to biases or even genuine diversity of the genetic effects in different populations. Here, we propose methods for estimating the required sample size to replicate an association across many studies with different amounts of between-study heterogeneity, when data are summarized through metaanalysis. We demonstrate thresholds of between-study heterogeneity (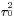 ) above which one cannot reach adequate power to replicate a proposed association at a specified level of statistical significance when k studies are performed (regardless of how large these studies are). Based on empirical evidence from 91 proposed gene–disease associations (50 on candidate genes and 41 from genome-wide association efforts), the observed between-study heterogeneity is often close to or even surpasses nonreplicability thresholds. With more modest between-study heterogeneity, the required sample size increases considerably compared with when no between-study heterogeneity exists. Increases are steep as is approached. Therefore, some true associations may not be practically possible to replicate with consistency, no matter how large studies are conducted. Efforts should be made to minimize between-study heterogeneity in targeted genetic effects. ) above which one cannot reach adequate power to replicate a proposed association at a specified level of statistical significance when k studies are performed (regardless of how large these studies are). Based on empirical evidence from 91 proposed gene–disease associations (50 on candidate genes and 41 from genome-wide association efforts), the observed between-study heterogeneity is often close to or even surpasses nonreplicability thresholds. With more modest between-study heterogeneity, the required sample size increases considerably compared with when no between-study heterogeneity exists. Increases are steep as is approached. Therefore, some true associations may not be practically possible to replicate with consistency, no matter how large studies are conducted. Efforts should be made to minimize between-study heterogeneity in targeted genetic effects.
Keywords: genome | heterogeneity | metaanalysis | polymorphism
Lack of replication of proposed gene–disease association has been seen repeatedly in the literature (1–5). Nonreplication often means that the original research findings reflected false positives. Replication is now considered a sine qua non for the rigorous documentation of proposed associations, and this is becoming even more prominent in the era of genome-wide association studies (6, 7). Nonreplication of a proposed association may be the desirable outcome in some situations, whereas it may be an error in others. For example, failure to replicate an association that arose because of genotyping error in the original study is desirable, whereas failure to replicate because of genotyping error in the replication study is wrong. Occasionally, nonreplication may occur even when a genuine association does exist and even if random measurement error is not large. The results of replicating studies may vary among themselves if biases (any systematic source of error, excluding random measurement error due to chance alone) affect differently the observed effects across various studies. Nonreplication may also arise if there is genuine diversity of the genetic effects in different populations and settings.
These situations may not be uncommon. Common biases include population stratification, misclassification of phenotype, genotyping error, and selection biases affecting the whole field of research, e.g., publication and selective reporting biases (8–11). Genuine differences in the genetic effects include differential linkage disequilibrium of the identified genetic marker with the true functional culprit gene variant in different populations (12); and association with a different, correlated phenotype. Differential linkage disequilibrium may be common in genome-wide association studies, because the tag polymorphisms are not selected based on functional evidence. We also are starting to see examples of associations for correlated phenotypes. For correlated phenotypes, failure of replication is desirable, because it points out that we need to search for an association with a different phenotype rather the one that was originally proposed. For example, an FTO variant showed heterogeneous associations in genome-wide association studies of diabetes (13), but it had a consistent association with body mass index and obesity across many studies (14). Some of the diabetes studies had matched cases and controls for body mass index, so no association was observed with diabetes, whereas in other studies the diabetic cases tended to be more obese than the controls. Finally, latent population-specific gene–gene or gene–environment interactions may result in different average genetic effects in different settings (15).
Here, we propose methods for estimating the required sample size to replicate an association across many studies with different amounts of between-study heterogeneity when data are summarized through metaanalysis (16–19). We performed simulations for which we assumed that a certain proposed gene–disease association would be tested in many different studies, and the data would then be synthesized by metaanalysis. Metaanalysis is the final step in asserting the credibility of effects (17–21). Metaanalysis across diverse populations also has become the standard for confirming proposed associations after massive genome-wide association testing (6, 7). We aimed to estimate what the total required sample size would be, depending on the frequency of the minor genetic variant of interest, the magnitude of the average genetic effect [odds ratio in multiplicative (log-additive) model], and the extent of heterogeneity (diversity) in the genetic effects across the different studies.
There are several different metrics for expressing between-study heterogeneity (22–25). Our simulations assumed different values of heterogeneity expressed by the between-study variance  . Calculations used random-effects models and the DerSimonian and Laird estimator of between-study variance (26). These models assume that the genetic effects are different across the different study populations, and they try to estimate the average population effect and the dispersion thereof (heterogeneity). . Calculations used random-effects models and the DerSimonian and Laird estimator of between-study variance (26). These models assume that the genetic effects are different across the different study populations, and they try to estimate the average population effect and the dispersion thereof (heterogeneity).
Results
Nonreplicability Thresholds. As demonstrated in detail in Methods (see also ref. 27), the required sample size n to detect an overall association with power (1–β) at a significance level of  when there are k studies and each one of them has a portion when there are k studies and each one of them has a portion 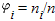 of the total sample can be estimated through of the total sample can be estimated through
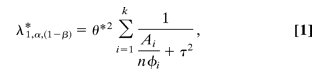
where 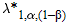 is the noncentrality parameter corresponding to a noncentral is the noncentrality parameter corresponding to a noncentral  2 variable that exceeds the upper percentile of the 2 distribution (1 – β)% of the time; 2 variable that exceeds the upper percentile of the 2 distribution (1 – β)% of the time; 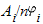 is the variance of the log odds ratio, where Ai is given by 1/[f1i(1 – f1i)] + 1/[f2i(1 –f2i)], with f1i and f2i being the frequencies of the genetic variant in controls and cases, respectively, of study i (i = 1, 2 , ... , k); and is the variance of the log odds ratio, where Ai is given by 1/[f1i(1 – f1i)] + 1/[f2i(1 –f2i)], with f1i and f2i being the frequencies of the genetic variant in controls and cases, respectively, of study i (i = 1, 2 , ... , k); and  * is the mean normalized genetic effect (log odds ratio). Under different assumptions for * , , Ai, and * is the mean normalized genetic effect (log odds ratio). Under different assumptions for * , , Ai, and 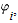 , one can iteratively find the sample size, n, that satisfies Equation 1. , one can iteratively find the sample size, n, that satisfies Equation 1.
For simplification, we consider that all k replication studies have the same sample size; i.e., 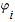 is the same for all studies. For a metaanalysis of k studies with increasing sample sizes and for common variants, in Equation 1 approaches zero, and thus we are left with is the same for all studies. For a metaanalysis of k studies with increasing sample sizes and for common variants, in Equation 1 approaches zero, and thus we are left with
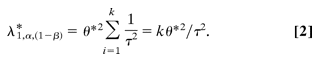
This result shows that cannot exceed 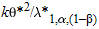 and that the equality holds when the total sample size approaches infinity. In other words, no sample size, no matter how large, would be sufficient to achieve the required power (1 – β) for the test for overall association if the between-study heterogeneity exceeds the threshold and that the equality holds when the total sample size approaches infinity. In other words, no sample size, no matter how large, would be sufficient to achieve the required power (1 – β) for the test for overall association if the between-study heterogeneity exceeds the threshold
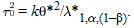
For example, when = 0.05 and (1 – β) = 0.80, we can use the CNONCT function in SAS to calculate the value of , which is equal to 7.849. To detect a log odds ratio * = 0.336 (corresponding to an odds ratio of 1.4) for k = 10 studies, has to be <0.14. With higher levels of between-study heterogeneity, power to overall replicate the association in the final metaanalysis of all data remains <80%, even at the very liberal = 0.05, no matter how large these 10 studies are. The decreases further when we ask for more stringent levels of statistical significance, and it reaches a value of 0.030 when we require genome-wide levels of significance ( = 0.0000001) to accept an association under otherwise similar *, β, and k.
Another useful metric is h0, which is defined as the ratio of  0/|*|; i.e., it states the largest allowed proportion of the effect size that the between-study deviation may represent, so that an association would still be detectable when the k replicating studies are combined. The h0 threshold is independent of the effect size. Table 1 shows the values of this threshold for different levels of (0.05, 0.01, 0.0001, and genome-wide 0.0000001) and for different levels of requested power. As shown, once we request genome-wide significance, the threshold changes relatively little for power between 50% and 95%. The h0 is linearly proportional to the square root of the number of studies k. For 10 studies, the h0 varies between 0.454 and 0.594, suggesting that nonreplicability ensues when the between-study standard deviation is about half of the effect. Conversely, with as many as 50 studies, the h0 varies between 1.011 and 1.329, suggesting that the nonreplicability threshold becomes more remote and will not ensue unless the between-study standard deviation is at least as large or larger than the full size of the effect. 0/|*|; i.e., it states the largest allowed proportion of the effect size that the between-study deviation may represent, so that an association would still be detectable when the k replicating studies are combined. The h0 threshold is independent of the effect size. Table 1 shows the values of this threshold for different levels of (0.05, 0.01, 0.0001, and genome-wide 0.0000001) and for different levels of requested power. As shown, once we request genome-wide significance, the threshold changes relatively little for power between 50% and 95%. The h0 is linearly proportional to the square root of the number of studies k. For 10 studies, the h0 varies between 0.454 and 0.594, suggesting that nonreplicability ensues when the between-study standard deviation is about half of the effect. Conversely, with as many as 50 studies, the h0 varies between 1.011 and 1.329, suggesting that the nonreplicability threshold becomes more remote and will not ensue unless the between-study standard deviation is at least as large or larger than the full size of the effect.
|
TABLE 1: Nonreplicability thresholds for different values of type I and II errors and for different number of studies |
Heterogeneity in Proposed Associations. The estimated 0 and h0 thresholds are not very high. Across 50 genetic associations proposed in the candidate gene era that reached nominal statistical significance (P < 0.05) in metaanalyses of all of the available data (28), 38 had different from zero. Figure 1a shows the distribution of and the distribution of h =  /|| (the ratio of the between-study study variation over the absolute effect size) in these 38 metaanalyses. The median values are 0.26 and 0.84. These values are on the high side of the range of thresholds of nonreplicability that we have estimated in Table 1, even for relatively lenient levels of statistical significance. Therefore, for several gene–disease associations, the high power to replicate them may not be reached, no matter how large the studies that we conduct are. Paradoxically, these associations would be true, but nonreplicable, if between-study heterogeneity remains in the range observed for postulated associations in the past. /|| (the ratio of the between-study study variation over the absolute effect size) in these 38 metaanalyses. The median values are 0.26 and 0.84. These values are on the high side of the range of thresholds of nonreplicability that we have estimated in Table 1, even for relatively lenient levels of statistical significance. Therefore, for several gene–disease associations, the high power to replicate them may not be reached, no matter how large the studies that we conduct are. Paradoxically, these associations would be true, but nonreplicable, if between-study heterogeneity remains in the range observed for postulated associations in the past.
We also estimated the values of and h for the 10 loci that have been considered to be "confirmed" susceptibility loci in a recent prospective metaanalysis of three genome-wide association studies of type 2 diabetes (29). For six of these studies, there was some between-study heterogeneity, with ranging between 0.017 and 0.138 and h ranging between 0.12 and 0.62 (Figure 1a). Although these values are smaller than those observed in the metaanalyses of published data from the candidate gene era, they still remain considerable and may interfere with the replicability of specific associations. Another recent genome-wide association study of breast cancer provided summary odds ratios for 31 polymorphisms that had been selected for further replication in 23 case-control studies (30). Eleven of these 31 polymorphisms have nominal P values of <0.05 by random-effects calculations. Of the 11 studies, five had = 0, whereas in the other six polymorphisms ranged from 0.028 to 0.075 and h ranged from 0.14 to 1.70 (Figure 1a). For the 20 "nonreplicated" breast cancer polymorphisms (P > 0.05 for the summary effect), only two had estimated = 0; for 12 polymorphisms, ranged from 0.013 to 0.1 and h ranged from 1.04 to 5.37; and in the other six, ranged from 0.027 to 0.14 and h ranged from 6.95 to 42.38 (Figure 1b). The summary effect sizes for these 20 nonreplicated polymorphisms were generally very small (corresponding to odds ratios of 0.95–1.04), but one cannot rule out completely the possibility that some of them may still mirror true associations but were not replicated because the heterogeneity was too much given the genetic effect sizes. [Supporting Information (SI) Table 2].
Estimates of Required Sample Size in the Presence of Heterogeneity. One may also estimate the required sample sizes to detect an association in the presence of more modest between-study heterogeneity (below the nonreplicability thresholds). These sample sizes can be compared against the respective sample sizes in the absence of any between-study heterogeneity. We considered a range of plausible values for the overall average genetic effect, corresponding to odds ratios of 1.05, 1.1, 1.2, 1.3, 1.4, and 2.0. We also considered a range of plausible values for that would be below the respective nonreplicability threshold given the specified odds ratio, = 0.0000001 and power of 80%. These thresholds are 0.0006, 0.0024, 0.0087, 0.0181, 0.0298, and 0.1263 for odds ratios of 1.05, 1.1, 1.2, 1.3, 1.4, and 2.0, respectively. We show results for 10 replicating studies of equal sample size that we conducted under the assumption that different numbers of studies would not change the results considerably. The simulations involved generating 10 values of i from a normal distribution N(*, ) for a hypothetical metaanalysis of 10 studies (i = 0.1). We considered a range of minor genetic variant frequencies in the controls, f1(0.05, 0.1, 0.2, 0.3, and 0.4). For each of the scenarios based on different minor genetic variant frequency, odds ratio, and , 10,000 simulations were carried out.
For an illustration of our simulation, SI Figure 3 gives the distributions of sample size obtained for = 0.002 and 0.007, respectively, when the odds ratio is 1.2 and the genotype frequency is 0.2. The mean estimated sample size for = 0.002 is 19,688 and the 95% confidence interval is given by 19,688 ± 190 whereas the mean sample size for = 0.007 is 76,449 and the 95% confidence interval is given by 76,449 ± 1,376. Mean estimated sample sizes are described from now on.
As shown in Figure 2, as expected, the required sample size requirement increased steeply with decreasing odds ratios and decreasing frequencies of the genetic variant. For example, when the odds ratio is 1.4, genetic variant frequency is 0.1, and there is no between-study heterogeneity ( = 0, essentially a fixed-effect model), the required sample size is 8,668. For the same genetic variant frequency and = 0 the required sample size increases to 362,298 when the odds ratio is 1.05. When the odds ratio is 1.2 and when = 0, the required sample size increased by 439% for a genetic variant frequency of 0.05 compared with a genetic variant frequency of 0.4. Similar trends can be seen for different values of .
For the same combination of genetic variant frequency and odds ratio, the required sample size also increases steeply with increasing values of , especially as approaches the threshold (Figure 2). For example, when the odds ratio is 1.2, for = 0, the required sample size ranged from 9,755 to 52,534 for the genotype frequencies considered; for = 0.002, the required sample size still ranged from 12,656 to 68,148. When = 0.007, the required sample size increased steeply (range from 49,126 to 264,630). As approached the = 0.0087 threshold, the required sample size tended to infinity. The same steep increase is documented for otherwise similar settings, but with = 0.05, in SI Figure 4.
Discussion
Our simulations show that some true associations may be nonreplicable; i.e., when many studies are conducted, the power to replicate the associations may remain below a given level, regardless of how large study populations we can amass in the replication efforts. This should not be seen as an argument that large-scale replication of proposed associations should not be pursued and intensely so. The field of human genome epidemiology has seen a gradual transformation from a domain of small, poorly replicated studies of single candidate genes (31) to massive testing with genome-wide platforms and extensive replication even upon the first publication of a postulated association (13, 29, 32–34). Replication sample sizes have gradually increased to exceed 40,000 subjects in some studies (14, 30). Our calculations suggest that such sample sizes or even larger are absolutely essential in generating sufficient power that a proposed association of small or modest effect size can be properly replicated, at least when between-study heterogeneity is not large. The very large sample sizes required also offer support to efforts to generate large-scale consortia (35) as well as biobanks (36) and new large-scale population cohorts (37), especially for research where case-control sampling is not feasible or appropriate.
We should caution that inferences for the presence or absence of an association are typically made based on some threshold, and, here, we have assumed frequentist thresholds (P values). Obviously, one should also examine the uncertainty in the summary estimate as conveyed by the confidence intervals. When the confidence intervals do not exclude large effects, more evidence from additional samples is likely to be sought trying to obtain a more conclusive answer. However, as we show, above the nonreplicability threshold, even with more data, the threshold of significance may still not be passed.
One might argue that we can raise the and h0 nonreplicability thresholds by performing more large studies (increasing k). However, this is only an artificial relief that does not hold in practice. The number of studies that can be performed is usually limited by the number of investigative teams working on a specific topic, and it is very uncommon that more than a dozen teams or so can put forth very large data sets in any field, including genetic associations. Splitting the data from a single team to many (sub)studies also is misleading: Point estimates in small substudies would have very large uncertainty; thus, seeming homogeneity would reflect simply lack of power to detect heterogeneity.
We should acknowledge that our sample size calculations assume for each time the same frequency for the genetic variant of interest across the different studies. Therefore, we consider populations with similar genetic background regarding the specific variant. When the genetic variant frequency varies across populations, those populations are likely to be even more heterogeneous; e.g., they may have different ethnic or racial descent. Preliminary evidence suggests that differences in genetic frequencies across populations of different racial descent usually are not accompanied by differences in the population-specific genetic effects (odds ratios) (38). Nevertheless, all other aspects being equal, considering populations with heterogeneous frequencies is likely to introduce more between-study heterogeneity, if anything, potentially leaving room for even less heterogeneity from other sources to reach the thresholds.
Our findings imply that the success of the replication process is contingent on efficiently reducing the between-study heterogeneity in the genetic effect in the replication studies. Reducing between-study heterogeneity may sometimes be feasible if the heterogeneity is due to errors and biases that can be amended with proper attention to study design and methodological issues. Such errors and biases include phenotype and genotype misclassification (39), population stratification (40), and selective reporting biases (41). Modest decreases in these sources of heterogeneity may allow the data to be brought to sufficient consistency, avoiding proximity to the nonreplicability threshold. Prospective metaanalysis of genome-wide association studies benefit from greater attention to genotyping and population stratification control (principal component analysis) and a lack of selective reporting problems.
Genuine heterogeneity also may be reduced by identifying the culprit genetic variant through fine mapping, sequencing, and functional studies for variants in the region of the markers that emerge from genome-wide testing (42). When heterogeneity is due to differential linkage disequilibrium of the unknown culprit marker in different populations, failure to replicate is desirable, because we realize that the identified marker has no generalizability for use as a prognostic test across different populations. The information is still useful from a biological perspective, e.g., pointing to a genetic area that needs more study. Tackling population-specific gene–gene and gene–environment interactions may be difficult at the current stage (43). Finally, if racial descent or some other population characteristic (e.g., gender) is considered to underlie the between-study heterogeneity, then the evaluation and synthesis of data on genetic effects should be performed separately for different subgroups. However, such a choice then needs to be supported with data that document the genetic subgroup differences. To date, such documentation is the exception (38, 44).
Eventually, even with the best efforts to minimize between-study heterogeneity, a sizeable proportion of genuine associations may remain spuriously nonreplicated. Our simulations provide evidence for an unavoidable uncertainty component in rejecting postulated associations.
Methods
Sample Size and Power Calculations for Metaanalysis: Conceptual Issues. Traditionally metaanalyses have been conducted retrospectively combining data from past studies, which leaves considerable room for biases. In addition, it may be argued that sample size and power calculations are not meaningful for retrospective data (45): Sample size has already been accrued and effects and their uncertainty have been observed. However, in the current setting of searching for gene–disease associations, replication is increasingly envisioned as a prospective effort. Typically, massive testing yields promising signals for specific polymorphisms that then have to be replicated. Several replication studies are often published in the same paper as the original discovery data set. The conduct of replicating studies can be seen as a prospective metaanalysis. In this setting, arguments against sample size calculations in metaanalysis are not valid.
Hedges and Piggott (27) have described procedures to compute statistical power of fixed- and random-effects tests of the mean effect size and tests for heterogeneity of effect size parameters across studies. We expand these methods here to calculate the required sample size for a prospective metaanalysis of replicating studies in the absence or presence of between-study heterogeneity.
Fixed- and Random-Effects Assumptions. Estimating an overall effect size 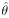 for a metaanalysis of k separate genetic association studies involve averaging the estimated effect size, for a metaanalysis of k separate genetic association studies involve averaging the estimated effect size, 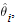 , of the of true effect size , of the of true effect size
i (i = 1, 2, ..., k) over all of the studies. For example, i could be the observed log odds ratio, log relative risk, risk difference, or mean difference (for continuous traits) in the ith case-control study designed to detect an association between a genetic variant and a complex disease. In the fixed-effect approach, homogeneity of the true effect sizes across studies, i.e., 1 = 2 = ... = k, is assumed. The overall effect size is then estimated as a weighted average,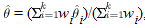 , where wi is the weight given to the ith case-control study. We can assume that i is approximately normally distributed with mean i and variance , where wi is the weight given to the ith case-control study. We can assume that i is approximately normally distributed with mean i and variance 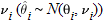 . Under this assumption, ~ N(, v), where . Under this assumption, ~ N(, v), where 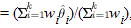 and 1/v = and 1/v =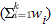 . Assuming equal sample size allocation for cases and controls, . Assuming equal sample size allocation for cases and controls, 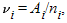 , where ni is the sample size for cases (or controls), and Ai depends on the type of effect size estimate. For example, if the effect size estimate is a log odds ratio, , where ni is the sample size for cases (or controls), and Ai depends on the type of effect size estimate. For example, if the effect size estimate is a log odds ratio,
Ai = 1/[f1i(1 – f1i )] + 1/(f2i (1 – f2i)], where f1i and f2i are the frequencies of the genetic variant in controls and cases, respectively, of the ith case-control study, i = 1, 2, ..., k.
When heterogeneity is present, the random effects model incorporates between-study variability into the overall estimate of the effect size. The estimate of effect size, , from the ith case-control study is assumed to have a
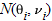 distribution as in a fixed-effect model, whereas the true effect sizes from individual studies, , are assumed to have a distribution as in a fixed-effect model, whereas the true effect sizes from individual studies, , are assumed to have a 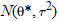 distribution, where is the between-study variance. Similar to the fixed-effect model, an overall estimate of random-effect sizes, *, is obtained by a weighted average of the effect sizes in individual case-control studies. The weight of the ith study in a random-effects metaanalysis, w*i, is given by distribution, where is the between-study variance. Similar to the fixed-effect model, an overall estimate of random-effect sizes, *, is obtained by a weighted average of the effect sizes in individual case-control studies. The weight of the ith study in a random-effects metaanalysis, w*i, is given by 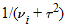 . Thus, the weight given to a study in random-effects metaanalysis depends not only on the variance of the effect size for that study but also on the heterogeneity between studies. As in the fixed-effect model, . Thus, the weight given to a study in random-effects metaanalysis depends not only on the variance of the effect size for that study but also on the heterogeneity between studies. As in the fixed-effect model, 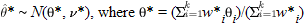 and and 
Here we have used random-effects calculations. Many genetic association studies and their replication efforts use fixed-effects analyses. However, the basic assumption of fixed effects is violated when there is any between-study heterogeneity. Fixed effects may hint to important genetic variability at a locus, but they generate inappropriately tight confidence intervals and low P values in the presence of heterogeneity (46, 47).
Furthermore, in some circumstances effect sizes and heterogeneity may be related to study sample size. For example, larger studies may include more diverse populations and/or a wider spectrum of disease, and such studies may be performed by more experienced investigators with lower error rates. However, these possibilities need to be examined empirically on a case by case basis.
Tests for Overall Association. Under the null hypothesis of no overall association, *2/v* has an approximately 2 distribution with one degree of freedom. Under the alternative hypothesis, *2/v* has a noncentral 2 distribution with one degree of freedom and noncentrality parameter 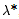 , given by , given by 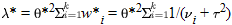 . .
Sample Size Estimation. Let  , where n is the total sample size for the k studies. Then , where n is the total sample size for the k studies. Then 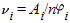 and the total sample size, n, required to detect an overall association with power (1 – β) at a significance level of is given by and the total sample size, n, required to detect an overall association with power (1 – β) at a significance level of is given by
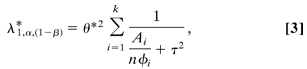
where 1,,1–β is the noncentrality parameter corresponding to a noncentral 2 variable that exceeds the upper percentile of the 2 distribution (1 – β)% of the time. Assuming that *, ,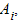 , and , and 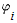 are known, one can iteratively find the approximate sample size, n, that satisfies Equation 1. are known, one can iteratively find the approximate sample size, n, that satisfies Equation 1.
Thresholds for Heterogeneity. For a metaanalysis on a common variant including k studies with increasing sample sizes and assuming that remains a constant, 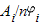 in Equation 1 approaches zero, and we are left with in Equation 1 approaches zero, and we are left with
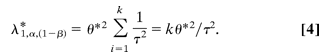
When the total sample size approaches infinity, the weights for a fixed-effect model tends to infinity, but the weights for a random-effects model tends to 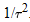 . This result shows that has to be less than or equal to . This result shows that has to be less than or equal to 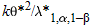 and that the equality holds when the total sample size approaches infinity. and that the equality holds when the total sample size approaches infinity.
Empirical Data from 91 Postulated Gene–Disease Associations. Data from 50 metaanalyses of gene–disease associations that reached nominal statistical significance (P < 0.05) with random-effects calculations have been published previously (28), and details on the literature searches and selection of genetic contrasts can be found elsewhere (1, 18, 28). Associations pertained to candidate gene polymorphisms and diverse disease phenotypes (no restriction set on disease phenotype). Data from prospective metaanalyses of 10 genetic variants implicated in type 2 diabetes by combining data from three genome-wide investigations along with their replication efforts are derived from Table 1 of Scott et al. (29). Each genome-wide association data set and its replication are considered as one study (29). Data from prospective metaanalyses of 31 genetic variants that were selected for further testing after successfully passing the first two screening stages of a genome-wide association on breast cancer are derived from the supplementary information of Easton et al. (30). The third-stage replication data include information from 23 studies. For each of these 91 postulated associations, we estimated the random-effects summary odds ratio and the DerSimonian and Laird estimator of the between-study variance to derive h. For the breast cancer postulated polymorphisms, we present separately those with nominally significant results (P < 0.05) versus those that did not reach nominal significance in the random effects metaanalysis.
Simulations and Software. Simulations were programmed by using the IML procedure in SAS Version 9 software.
Footnotes
¶To whom correspondence should be addressed. E-mail: jioannid@cc.uoi.gr
Author contributions: R.M., M.J.K., and J.P.A.I. designed research; R.M., T.L., and J.P.A.I. performed research; R.M., M.J.K., T.L., and J.P.A.I. analyzed data; and R.M. and J.P.A.I. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. B.S.W. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/cgi/content/full/0705554105/DC1.
© 2008 by The National Academy of Sciences of the USA
References
- Ioannidis JP, Trikalinos TA, Ntzani EE, Contopoulos-Ioannidis DG (2003) Lancet 361:567–571.
- Cardon LR, Bell JI (2001) Nat Rev Genet 2:91–99.
- Redden DT, Allison DB (2003) J Nutr 133:3323–3326.
- Hirschhorn JN, Lohmueller K, Byrne E, Hirschhorn K (2002) Genet Med 4:45–61.
- Hirschhorn JN, Altshuler D (2002) J Clin Endocrinol Metab 87:4438–4441.
- Ioannidis JP (2007) Hum Hered 64:203–213.
- Chanock SJ, Manolio T, Boehnke M, Boerwinkle E, Hunter DJ, Thomas G, Hirschhorn JN, Abecasis G, Altshuler D, Bailey-Wilson JE, et al. (2007) Nature 447:655–660.
- Clayton DG, Walker NM, Smyth DJ, Pask R, Cooper JD, Maier LM, Smink LJ, Lam AC, Ovington NR, Stevens HE, et al. (2005) Nat Genet 37:1243–1246.
- Evangelou E, Trikalinos TA, Salanti G, Ioannidis JP (2006) PLoS Genet 2:e123.
- Pan Z, Trikalinos TA, Kavvoura FK, Lau J, Ioannidis JP (2005) PLoS Med 2:e334.
- Calnan M, Smith GD, Sterne JA (2006) J Clin Epidemiol 59:1312–1318.
- Hirschhorn J, Daly MJ (2005) Nat Rev Genet 6:95–108.
- Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, Timpson NJ, Perry JRB, Rayner NW, Freathy RM, et al. (2007) Science 316:1336–1341.
- Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, Perry JRB, Elliott KS, Lango H, Rayner NW, et al. (2007) Science 316:889–894.
- Hunter DJ (2005) Nat Rev Genet 6:287–298.
- Lau J, Ioannidis JP, Schmid CH (1997) Ann Intern Med 127:820–826.
- Mosteller F, Colditz GA (1996) Annu Rev Public Health 17:1–23.
- Ioannidis JP, Ntzani EE, Trikalinos TA, Contopoulos-Ioannidis DG (2001) Nat Genet 29:306–309.
- Munafo MR, Flint J (2004) Trends Genet 20:439–444.
- Patsopoulos N, Apostolos AA, Ioannidis JP (2005) J Am Med Assoc 293:2362–2366.
- Lau J, Ioannidis JPA, Schmid CH (1998) Lancet 351:123–127.
- Higgins JPT, Thompson SG, Deeks JJ, Altman D (2002) J Health Serv Res Policy 7:51–61.
- Song F, Sheldon TA, Sutton AJ, Abrams KR, Jones DR (2001) Eval Health Prof 24:126–151.
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG (2003) Biol Mol J 327:557–560.
- Thompson SG, Higgins JPT (2002) Stat Med 21:1559–1573.
- DerSimonian R, Laird NM (1986) (1986) Contr Clin Trials 7:177–188.
- Hedges LV, Pigott DP (2001) Psychol Methods 6:203–217.
- Ioannidis JPA, Trikalinos TA, Khoury MJ (2006) Am J Epidemiol 164:609–614.
- Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, Erdos MR, Stringham HM, Chines PS, Jackson AU, et al. (2007) Science 316:1341–1345.
- Easton DF, Pooley KA, Dunning AM, Pharoah PDP, Thompson D, Ballinger DG, Struewing JP, Morrison J, Field H, Luben R, et al. (2007) Nature 477:1087–1093.
- Ioannidis JP (2003) Trends Mol Med 9:135–138.
- Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PIW, Chen H, Roix JJ, Kathiresan S, et al. Diabetes Genetics Initiative of Broad Institute of Harvard abd MIT, Lund University and Novartis Institutes for Biomedical Research (2007) Science 316:1331–1336.
- Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, Jonasdottir A, Sigurdsson A, Baker A, Palsson A, et al. (2007) Science 316:1491–1493.
- McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Rioberts R, Cox DR, Hinds DA, Pennacchio LA, Tybiaerg-Hansen A, Folsom AR, et al. (2007) Science 316:1488–1491.
- Seminara D, Khoury M, O'Brian TR, Manolio T, Gwin ML, Little J, Higgins JPT, Bernstein JL, Boffetta P, Bondy M, et al. (2007) Epidemiology 18:1–8.
- Ioannidis JPA (2007) J Epidemiol Community Health 61:757–758.
- Collins FS, Manolio T (2007) Nature 445:259.
- Ioannidis JPA, Ntzani EE, Trikalinos TA (2004) Nat Genet 36:1312–1318.
- Pompanon F, Bonin A, Bellemain E, Taberlet P (2005) Nat Rev Genet 6:847–859.
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D (2006) Nat Genet 38:904–909.
- Service S, DeYoung J, Karayiorgou M, Ross JL, Pretorious H, Bedoya G, Ospina J, Ruiz-Linares A, Macedo A, Palha JA, et al. (2006) Nat Genet 38:556–560.
- Greenland S (1983) Stat Med 2:243–251.
- Zumbo BD, Hubley AM (1998) Statistician 47:385–388.
- Patsopoulos NA, Tatsioni A, Ioannidis JP (2007) J Am Med Assoc 298:880–893.
- Hedges LV, Pigott DP (2004) Psychol Methods 9:426–445.
- Ioannidis JP, Patsopoulos NA, Evangelou E (2007) PLoS ONE 2:e841.
- Fleiss JL (1993) Stat Methods Med Res 2:121–145.
|

