|
About CDER - Report |
U.S. Food and Drug Administration • Center for Drug Evaluation and Research |
|
About CDER - Report |
|
|
CDER Report to the Nation: 2002 Table of Contents Adobe Acrobat version of this document Drug Review (continued)Index
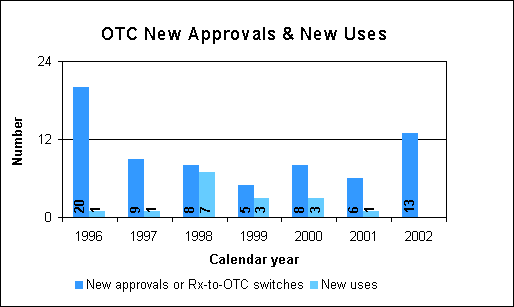
Over the Counter Drug Review
We approved 13 new drugs or Rx-to-OTC switches, 11 of which can be used by children. Among the approvals are:
First nonsedating antihistamine approved for OTC useLoratadine provides temporary relief of symptoms of hay fever or other upper respiratory allergies, runny nose, sneezing, itchy, watery eyes and itching of the nose or throat. We approved six formulations. The first three are for people age 6 and older, the syrup is for those as young as 2, while combinations with a decongestant are for those 12 and older.
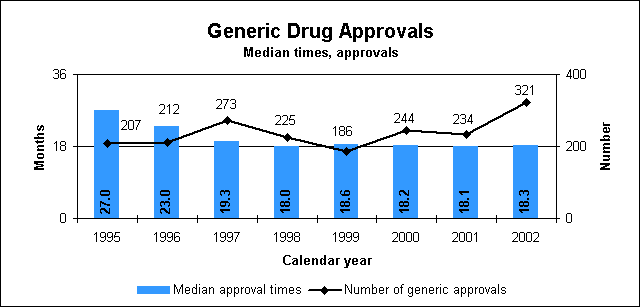
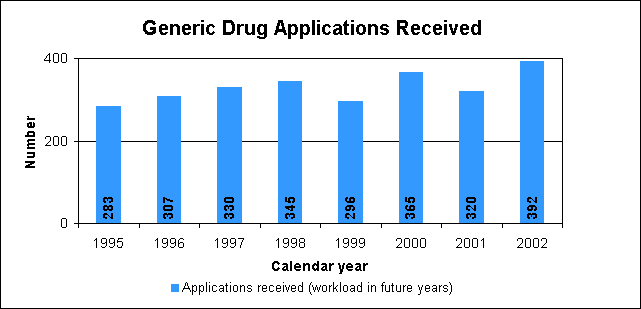
Improved labels for OTC medicinesAmerican consumers are benefiting from easy-to-understand labels on drugs they buy without a prescription. A mandatory changeover to the new labels, titled "Drug Facts," began in 2002. How we regulate OTC drugsWe publish monographs that establish acceptable ingredients, doses, formulations and consumer labeling for OTC drugs. Products that conform to a final monograph may be marketed without prior FDA clearance. Drugs can also be approved for OTC sale through the new drug review process. Generic Drug ReviewWe approved 321 generic drug products in 2002, including 80 products that represent the first time a generic drug was available for the brand-name product. The median approval time for generic drugs was 18.3 months. The median statistic for total approval time has hovered at about 18 to 19 months for five years. We are making changes to decrease the overall time to approval of applications. We are improving the efficiency of our generic drug review process and increasing the number of chemistry reviewers by one-third. Generic drugs a top priority"Encouraging rapid and fair access to generic medications after the expiration of appropriate patent protection" is one of the major priorities for new FDA Commissioner Mark McClellan, M.D., Ph.D. He noted that the generic drugs play an essential role in promoting the health of Americans. He assured the generic industry that we would work to reduce the time to approval for generic products and that generics must be safe and effective. We will work to identify steps such as improving guidance and communication to help improve the overall quality of applications thus gaining faster approvals. A copy of his remarks is available at http://www.fda.gov/oc/speeches/2002/gpha.html.
Generic drug statistics
Tentative approvals
Generic drug Web siteYou can find more information about our generic drug program at http://internet-dev.fda.gov/cder/ogd/index.htm. Notable 2002 generic drug approvalsExamples of first-time approvals for the brand-name equivalent drug are:
Our approval of generic versions of these drugs last year could save American consumers and the federal government hundreds of millions of dollars each year. We also issued 63 tentative approvals and 20 approvables last year:
Building consumer confidence in generic drugsWe launched our program to promote consumer confidence in the safety and effectiveness of generic drugs. We are informing health care practitioners and consumers about the rigorous review and approval process that generic drugs undergo before we approve them for sale in this country. Partnerships and networking with other groups are helping us to bring the message to more people. The consumer education program includes:
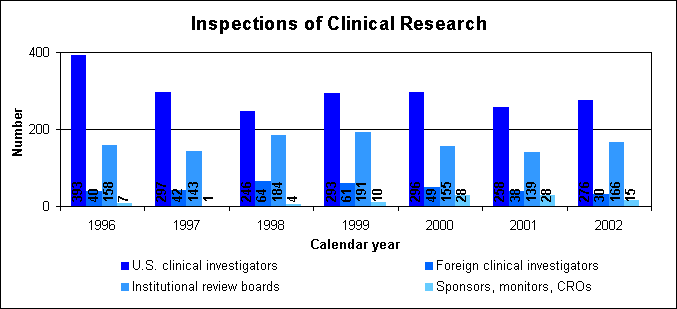
Our generic drug public service announcements are at http://www.fda.gov/cder/consumerinfo/generic_info/default.htm. Generic drug electronic submissionsThrough public presentations, we are encouraging the generic drug industry to submit their applications electronically. More information electronic submissions is below. How we approve generic drugsGenerics are not required to repeat the extensive clinical trials used in the development of the original, brand-name drug. Instead, they must show bioequivalence to the brand-name reference listed drug. Scientists measure the amount of the generic drug that reaches the bloodstream and how long it takes to get there. This rate and extent of absorption is called bioavailability. The bioavailability of the generic drug is then compared to that of the brand-name reference listed drug. The generic version must deliver the same amount of active ingredients into a patient's bloodstream and in the same time as the brand-name reference listed drug. Brand-name drugs are subject to the same bioequivalency tests as generics when their manufacturers reformulate them. Assessing Data Quality, Research RisksTo protect the rights and welfare of volunteers and verify the quality and integrity of data submitted for our review, we perform on-site inspections of clinical trial study sites, institutional review boards, sponsors, study monitors and contract research organizations. Our programs to protect volunteers are challenged by increases in the number of clinical trials; the types and complexity of products undergoing testing; and the increased number of trials performed in countries with less experience and limited or no standards for conducting clinical research. When obtaining data about the safety and effectiveness of drugs, sponsors rely on human volunteers to take part in clinical studies. Protecting volunteers from research risks is a critical responsibility for us and all involved in clinical trials, including manufacturers, institutional review boards, study sponsors, clinical investigators and their staffs, monitors, contract research organizations, hospitals and other institutions. Sponsors and clinical investigators protect volunteers by ensuring that:
Special attention is given to protecting vulnerable populations, such as children, the mentally impaired or prisoners. We require sponsors to disclose financial interests of clinical investigators who conduct studies for them. This helps identify potential sources of bias in the design, conduct, reporting and analysis of clinical studies.
Inspections of clinical research in 2002We conducted a total of 487 inspections of clinical research:
Top 5 deficiency categories for clinical investigator inspections
International inspections of clinical researchWe have conducted 490 inspections of clinical research in 51 countries from 1980 to 2002. We participate in international efforts to strengthen protections for human volunteers worldwide and encourage clinical investigators to conduct studies according to the highest ethical principles. These efforts include our work with the International Conference on Harmonization and the Declaration of Helsinki. User Fee ProgramAmericans deserve timely access to potentially lifesaving new drugs as soon as possible once they are proven safe and effective. The Prescription Drug User Fee Act of 1992 received its third five-year extension last year, known as PDUFA III. This reauthorization will ensure that we have the expert staff and resources to review applications promptly and get safe, effective new drugs into the hands of the people who need them. PDUFA III maintains the high review performance goals of PDUFA II, which included reduced drug review times and increased and accelerated our consultations with drug sponsors. In addition, PDUFA III remedies resource shortages that affected the program in recent years. Under PDUFA II, we collected significantly less in user fees than estimated due to a reduced number of new drug applications and an increased proportion of submissions whose fees were waived. The reauthorization puts the user fee program on a sound financial basis. We are also concerned about the safety of new medicines following approval. In recent years, 50 percent of all new drugs worldwide have been launched in the United States, and American patients have had access to 78 percent of the world's new drugs within the first year of their introduction. PDUFA III allows us to spend some user fees to increase surveillance of the safety of medicines during their first two years on the market or three years for potentially dangerous medications. It is during this initial period, when new medicines enter into wide use, that we are best able to identify and counter adverse side effects that did not appear during the clinical trials. Full information on PDUFA III, including the latest performance and procedure goals, is on the Web at http://www.fda.gov/oc/pdufa/PDUFA3.html. User fee performanceUnder legislation authorizing us to collect user fees for drug reviews, we agreed to specific performance goals for the prompt review of submissions.
Internet resources for user feesOur user fee Web site has links to more documents and information including our user fee performance report to Congress. The page is at http://www.fda.gov/cder/pdufa/default.htm. Electronic SubmissionsThe number of new drug applications submitted electronically continues to grow. Last year's electronic submissions were double the number submitted in the previous year. Overall, we had more electronic submissions last year than in the previous four years combined. The number of participating companies and the number of applications with electronic components continues to grow. About 70 percent of newly filed new drug applications have an electronic component, and two-thirds are completely electronic. About 17 percent of new or expanded use applications have an electronic component with 85 percent being completely electronic. Last year, we began receiving generic drug applications in electronic format. We continue to receive electronic drug advertising material in electronic format. Reviewers continue to find that electronic submissions provided as described in our guidance documents allow them to be more efficient. Our training programs include hands on classroom training as well as on-site training to teams receiving electronic applications. This training improves the ability to use the submissions effectively. We have been working with other regulatory agencies and pharmaceutical groups in the International Conference on Harmonization to complete the electronic common technical document. In 2003, we expect to begin receiving investigational new drug applications, annual reports and Drug Master Files in e-CTD format. In addition to receiving electronic applications, we have received over 22,000 electronic individual case safety reports from manufacturers. These reports are transmitted electronically and automatically entered into the Adverse Event Report System. This allows the reviewers to evaluate the reports sooner and reduces our resources for entering information into the system. Internet resourcesMore information on our electronic submissions program is at http://www.fda.gov/cder/regulatory/ersr/. Antimicrobial ResistanceThe emergence of drug-resistant bacteria is considered to be a major threat to the public health. We developed a regulation outlining new labeling designed to help reduce the development of drug-resistant bacterial strains. This rule became final in February 2003 and aims at reducing the inappropriate prescription of antibiotics to children and adults for common ailments such as ear infections and chronic coughs. Details of our other efforts and resources are at http://www.fda.gov/cder/drug/antimicrobial/default.htm. Public meeting on antimicrobial drug developmentWe along with industry and academia cosponsored a two-day public meeting to explore the scientific and regulatory issues in developing drugs to treat highly resistant pathogens. Pregnancy labelingWe have reviewed the current system of labeling drugs for use by pregnant women and are developing an improved, more comprehensive and clinically meaningful approach. We are consulting with multiple government agencies, medical experts, consumer groups and the pharmaceutical industry to develop this new labeling format. Last year, we began seeking public comment on a draft guidance that will provide sponsors with advice on how to establish pregnancy exposure registries. Registries that prospectively monitor the outcomes of pregnancies in women exposed to a specific drug can provide clinically relevant human data for treating or counseling patients who are pregnant or anticipating pregnancy.
FDA/Center for Drug Evaluation and Research |