|
About CDER - Report |
U.S. Food and Drug Administration • Center for Drug Evaluation and Research |
|
About CDER - Report |
|
|
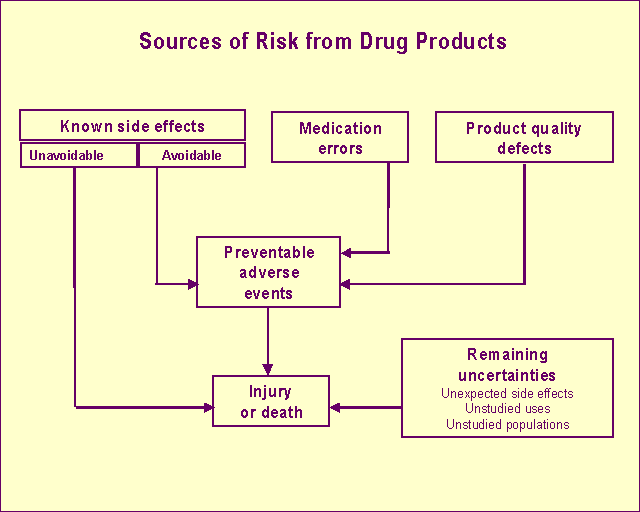
CDER Report to the Nation: 1999 Drug Safety and QualityThe practical size of premarketing clinical trials means that we cannot learn everything about the safety profile of a drug before we approve it. Therefore, a degree of uncertainty always exists about both the benefits and risks from drugs. The trade-off for accepting these uncertainties is our continued vigilance along with that of the industry to collect and assess data during the postmarketing life of a drug. We monitor the quality of marketed drugs and their promotional materials through product testing and surveillance. In addition, we develop policies, guidance and standards for drug labeling, current good manufacturing practices, clinical and good laboratory practices and industry practices to demonstrate the safety and effectiveness of drugs. MissionProtect the public health by ensuring that human drugs are safe and effective. Types of risks from medicinesProduct quality defects. These are controlled through good manufacturing practices, monitoring and surveillance. Known side effects. Predictable adverse events are identified in the drug's labeling. Known side effects cause the majority of injuries and deaths resulting from using medicines. There are avoidable and unavoidable side effects:
Medication errors. The drug is administered incorrectly or the wrong drug or dose is administered. Remaining uncertainties. These include unexpected side effects, long-term effects and unstudied uses and populations. For example, a rare event occurring in fewer than 1 in 10,000 persons won't be identified in normal premarket testing.
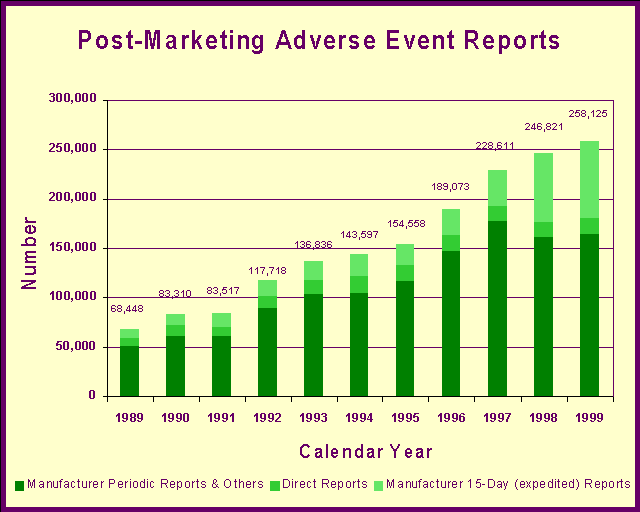
Risk managementFor a 164-page report on current and recommended premarket and postmarket risk assessment procedures and surveillance programs, last year's FDA report, Managing the Risks form Medical Product Use: Creating a Risk Management Framework, is available on the World Wide Web at http://www.fda.gov/oc/tfrm/riskmanagement.pdf. Drug SafetyWe evaluate the ongoing safety profiles of drugs available to American consumers using a variety of tools and disciplines. We maintain a system of postmarketing surveillance and risk assessment programs to identify adverse events that did not appear during the drug development process. We monitor adverse events such as adverse reactions, drug-drug interactions and poisonings. We use this information to update drug labeling and, on rare occasions, reevaluate the approval or marketing decision. Adverse event reportingLast year, we received 258,125 reports of suspected drug-related adverse events:
As we discover new knowledge about a drug's safety profile, we make risk assessments and decisions about the most appropriate way to manage any new risk or new perspective on a previously known risk. Risk management methods include new labeling, "Dear Health Care Practitioner" letters, restricted distribution programs or product marketing termination. We enforce regulations on adverse event reporting to assure that reports are accurate, timely and complete. During fiscal year 1999, there were 50 inspections of foreign and domestic firms for adverse event reporting. In addition, we gave guidance on policy issues and held meetings with industry to discuss their adverse event reporting practices. We approved four warning letters and three untitled letters citing adverse event reporting deficiencies. The 50 inspections in fiscal year 1999 compare to 51 conducted in fiscal year 1998, 33 in fiscal year 1997 and 17 in fiscal year 1996.
Report types
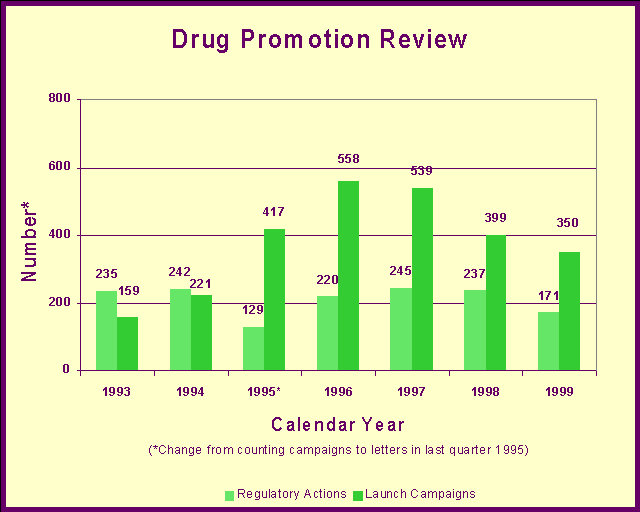
Information technologyA powerful tool for detecting signals is the computerized spontaneous reporting evaluation system. We have replaced our previous computerized system with a new, state-of-the-art system: the Adverse Event Reporting System. This system combines the voluntary adverse drug reaction reports from MedWatch and the required reports from manufacturers. These reports often form the basis of various "signals" that there may be a potential for serious, unrecognized, drug-associated events. After the signal is generated, further testing of the hypothesis is undertaken using various epidemiological and analytic databases, studies and other instruments and resources. The Adverse Event Reporting System offers paper and electronic submissions options, international compatibility and pharmacovigilance screening activities. MedWatchIn 1999, we took over administration of MedWatch under which health professionals and the public can voluntarily report serious reactions and problems with all FDA-regulated medical products. Reports can be filed by mail, fax, telephone or the Internet. The program enhances the effectiveness of postmarketing surveillance by rapidly identifying significant health hazards associated with them and notifying health professionals and the public of these hazards. We educate health professionals and consumers about the importance of recognizing and reporting serious adverse events and product problems, including medication errors. We rapidly disseminate safety information through the World Wide Web and by e-mail notification available to both to health professionals and the public. Our education program includes speeches, articles and exhibits. Medication errorsWe help ensure the safe use of drugs we approve by identifying and avoiding brand names that contribute to problems in prescribing, dispensing or administration of the product. Therapeutic inequivalence reportingWe identify and evaluate reports of therapeutic failures and toxicities that could indicate one produce is not equivalent to another similar product. Internet resourcesYou can learn more about the Adverse Event Reporting System at http://www.fda.gov/cder/aers/index.htm. The latest medical product safety information can be found on the MedWatch Website at http://www.fda.gov/medwatch/. You can sign up for immediate e-mail notification of MedWatch safety information at http://www.fda.gov/medwatch/new.htm. Drug Promotion ReviewThe information about a drug available to physicians and consumers is just as important to its safe use as drug quality. We promote and protect the health of Americans by ensuring that drug advertisements and other promotional materials are truthful and balanced. The Center operates a comprehensive program of education, surveillance and enforcement about drug advertising and promotion. In some instances, we review drug advertisements and other promotional materials before drug companies launch marketing campaigns that introduce new drugs or introduce new indications or dosages for approved drugs. In 1999, the Center issued 337 advisory letters to companies regarding their promotional materials for launch campaigns. We issued 171 regulatory action letters to pharmaceutical companies for prescription drug promotions determined to be false, misleading or lacking in fair balance. These were either "untitled" letters for minor violations or "warning" letters for serious or repeat violations. The Center also issued 773 other advisory, acknowledgment or closure letters to the industry regarding prescription drug promotional materials.
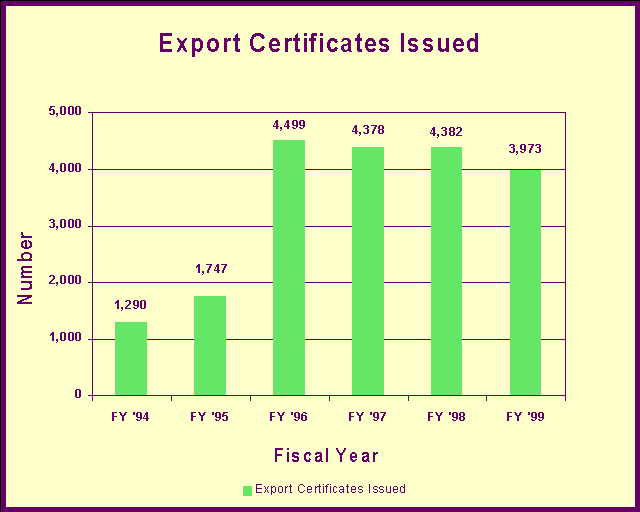
Direct-to-consumer advertisingWe issued 247 combined advisory and regulatory action letters regarding direct-to-consumer promotion. We issued a final guidance on direct-to-consumer broadcast advertisements that covered human and animal prescription drugs and human biologics. We completed a national telephone survey of patient attitudes and behaviors in relation to direct-to-consumer advertising, focusing on the patient-physician interaction. We also began research to examine how consumers interpret specific direct-to-consumer advertisements. Improved patient information for prescription drugsWe continued our research, education and outreach activities in support of the private plan to provide patients with useful information about their prescription drugs. We have been working with industry, nonprofit agencies and academic groups to ensure that 75 percent of patients receive useful information about their new prescriptions by the year 2000. We completed a study examining the current status of the private sector plan's progress toward achieving the year 2000 goal. We issued a final rule that requires FDA-approved patient labeling (medication guides) for especially risky prescription drug products. Risk vs. benefit communications researchWe are conducting research to assess the public's ability to understand risk and benefit information. The goal is to develop useful and meaningful ways of presenting important information about a drug's known risks and benefits. Export CertificatesWe promote goodwill and cooperation between the United States and foreign governments through the Export Certificate Program. These certificates enable American manufacturers to export their products to foreign customers and foreign governments. The demand for certificates by foreign governments remained high due to expanding world trade, ongoing international harmonization initiatives and international development agreements. The certificates attest that the drug products are subject to inspection by the FDA and are manufactured in compliance with current good manufacturing practices.
What export certificates doExport certificates verify that the drug products being exported:
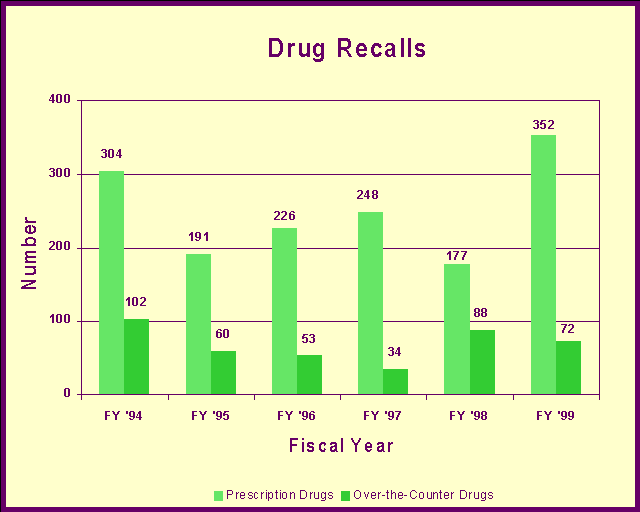
Drug Recalls and WithdrawalsWe coordinate drug recall information and prepare health hazard evaluations to determine the risk to public health by products being recalled. We classify recall actions in accordance to the level of risk, and we participate in determining recall strategies based upon the hazard and other factors including distribution patterns and market availability. We determine the need for public warnings and assist the recalling firm with public notification. Top 10 reasons for drug recalls in fiscal year 1999:
Voluntary recallsA recall is a voluntary action taken by a manufacturer or distributor to carry out their responsibility to protect the public health when they need to remove or correct a marketed drug product that presents a significant risk to public health. A voluntary recall is more efficient and effective in assuring timely consumer protection than an FDA initiated court action or seizure. In some cases, drugs are withdrawn from the market. Last year, manufacturers withdrew these two drugs for safety reasons:
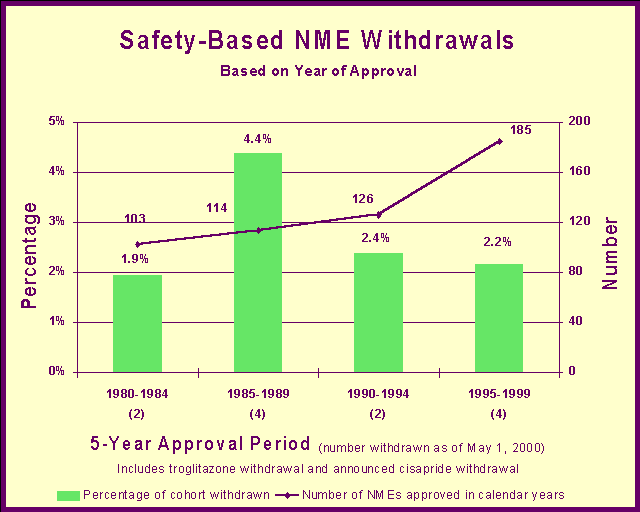
Safety-based NME withdrawalsThe record of withdrawal of drugs approved in recent years compares favorably to previous periods when we were criticized for taking too long to review drug applications. Nonetheless, the increased number of drugs and the large number of patients taking multiple drugs have created the potential for more drug safety problems. We are exploring these issues in a systematic manner with our partners in industry, academia, state and local governments, and patient and consumer organizations. Drug Product QualityWe provide comprehensive regulatory coverage of the production and distribution of drug products. This helps ensure that drugs are safe, effective and in compliance with applicable current regulations for good manufacturing practices. We manage inspection programs designed to minimize consumer exposure to defective drug products. We have two basic strategies to meet this goal:
We identify, evaluate and analyze inspection findings for trends. We develop guidances to assist drug manufacturers in gaining a better understanding of our regulations. We communicate the expectations of compliance through outreach programs. We review all international pharmaceutical inspection reports. We determine which foreign manufacturers are acceptable to supply active pharmaceutical ingredients or finished drug products to the U.S. market. Manufacturing plant inspectionsFDA field offices conduct inspections of plants that manufacture, test, package and label drugs. There are more than 18,000 of these plants in the United States. Before a drug is approved, FDA investigators must determine if data submitted in the firm's application are authentic and if the plant is in compliance with good manufacturing practices. After a drug is approved, FDA conducts an inspection to make sure a firm can consistently manufacture the product. Finally, routine inspections evaluate the firm's entire operations.
Reporting systems for drug quality problemsTwo important tools help us rapidly identify significant health hazards associated with the manufacturing and packaging of drugs:
Drug shortagesWe attempt to prevent or alleviate shortages of medically necessary drugs. We coordinated responses to 10 drug shortage situations in fiscal year 1999. These efforts assured the availability of necessary drug products for the treatment several serious and life-threatening diseases, such as myasthenia gravis, tuberculosis, AIDS and cancer. Surveillance sampling of drugsThe Drug Quality Surveillance Sampling Program helps determine the quality of imported and domestic drugs distributed in the United States. Samples of drug products are tested for conformance with quality specifications to ensure that the nation's drug supply is safe and effective and to provide rapid identification of emerging problems. We have intensified surveillance of imported drug products because of the increased number of imports. During fiscal year 1999, we surveyed 56 drug products. The analysis of 216 samples was complete as of May 1, 2000. Twenty samples failed various quality specifications including impurity tests, dissolution requirements, net content and the limit for free salicylic acid. Although investigational follow-up is in-process for some samples, compliance achievements to date include packaging and manufacturing revisions and other recommendations. Sampling criteriaWe chose drugs for the sampling program based on the following criteria:
Unsubstantiated claims, fraudulent and hazardous productsWe encounter many products that are vitamins, minerals, amino acids and herbal preparations with labeled drug claims. These products may be labeled as dietary supplements but make claims that they are safe and effective for the prevention, treatment or cure of such diseases as AIDS or cancer. Because these claims are unsubstantiated, they could present a health hazard when consumers delay or avoid seeking appropriate medical care.
Party drugs seizedThe so-called "party drugs" GHB (gamma-hydroxybutyrate), GBL (gamma-butyrolactone) and 1-4 butanediol are examples of ingredients that have been promoted for illicit activities including date rape. We have seized products that contain these ingredients because they pose a significant risk of injury or death.
FDA/Center for Drug Evaluation and Research |