|
 |
 |
|
 |
FDA Home Page | Search FDA Site | FDA A-Z Index | Contact FDA

 |
 |
Printable version of this report [568 KB PDF]
This page contains links to documents in Portable Document
Format (PDF). To view or
print these documents, you must use the Adobe Acrobat viewer.
Acrobat
is free and available directly from Adobe's website with full installation
instructions.
Report on FY 2005 and FY 2006 PDUFA Goals
Original Applications
Resubmitted Applications
Efficacy Supplements
Resubmitted Efficacy Supplements
Manufacturing Supplements
Report on Other FY 2006 PDUFA Goals, Initiatives, and Commitments
Meeting Management
Special Protocol Assessments
Response to Clinical Holds
Major Dispute Resolutions
First Cycle Filing Review Notification
Reviewable Unit Letter Notification
PDUFA III Management Initiatives Accomplishments
Electronic Applications and Submissions Accomplishments
Appendices
Appendix A: PDUFA Performance Goals, FY 2003 - FY 2007
Appendix B: List of Approved Applications
I am pleased to present the Food and Drug Administration's (FDA's) fiscal year (FY) 2006 Performance Report to the President and Congress for the Prescription Drug User Fee Act (PDUFA). This report marks the 14th year of PDUFA, and completion of the 4th year of the most recent 5-year reauthorization (PDUFA III). Resources provided to FDA under PDUFA legislation have been instrumental in new drugs reaching consumers in a timelier manner.
PDUFA I (FY 1993 through FY 1997) challenged FDA with goals to speed FDA review of new drug applications (NDAs) and biologics licensing applications (BLAs) without compromising safety. Over the course of PDUFA I, FDA exceeded all of its review performance goals. PDUFA II (FY 1998 through FY 2002) added goals to improve the process of new drug development before submission of the NDA or BLA. Under PDUFA II, most review times were shortened and FDA met or exceeded nearly all its review performance goals.
PDUFA III (FY 2003 through FY 2007) expanded fee funding to support FDA postmarket risk management and established several initiatives to improve application submissions and FDA-sponsor interactions during drug development and application review. It is believed that early and more frequent consultation with FDA may help sponsors improve the quality of their drug development and related applications. In this area, FDA continues to experience significant and unanticipated increases in company requests for meetings and special protocol assessments that began when the PDUFA procedural and processing goals were instituted during PDUFA II. While these FDA-sponsor interactions are important to improving drug quality, they also impose a substantial amount of additional work for FDA. FDA continues to meet or exceed most review performance goals, including exceeding the goal for reviewing priority New Molecular Entities (NMEs) within 6 months, but fell 1 percent short of the 90 percent on-time review goal for priority NDAs and BLAs. Although FDA's review performance for special protocol assessment requests improved from the previous year and exceeded the FY 2006 goal, FDA was not able to meet performance targets for other procedural and processing goals. However, FDA made progress in its PDUFA III management initiatives, met or exceeded most FY 2005 goals, and is exceeding all FY 2006 goals.
With PDUFA III expiring in September 2007, the reauthorization of PDUFA is essential to maintain the resources required to sustain the advances made in FDA review performance and to continue to advance biomedical progress.
Andrew C. von Eschenbach, M.D.
Commissioner of Food and Drugs
This report presents FDA’s performance in meeting annual PDUFA review goals. Review performance for applications and submissions received in FY 2005 and initially reported in the FY 2005 report is updated and finalized. FDA’s progress in meeting the quantifiable PDUFA review performance goals for FY 2005 and FY 2006 submissions and the FY 2006 procedural and processing goals are covered in this report. Additionally, this report describes FDA’s progress in accomplishing new management initiatives and in meeting the information technology commitments of PDUFA III.
Workload related to review processes increased in most categories from FY 2005 to FY 2006. The number of original NDAs and BLAs increased by 10 percent and the number of NDA and BLA efficacy supplements increased by 15 percent. FDA reviewed and acted on all but two of the original applications submitted during FY 2005 and, as of September 30, 2006, FDA met or exceeded almost all of the review performance goals. The single review goal not met in FY 2005 was to review and act on 90 percent of priority NDAs (which include NMEs) and BLAs within 6 months; the FDA performance level for this goal was 89 percent. FDA can now report that in FY 2005 it:
Preliminary review performance for FY 2006 indicates FDA is meeting or exceeding all on-time performance goals for applications and resubmissions reviewed and acted on as of September 30, 2006. FDA is also meeting or exceeding all PDUFA III management initiative performance goals for FY 2006.
Workload related to the procedural and processing goals moderately increased again in FY 2006 with higher numbers in meeting requests (up 5 percent from FY 2005), meetings scheduled (up 3 percent from FY 2005), special protocol assessments (up 2 percent), and clinical hold responses (up 14 percent from FY 2005). These increases affect the same FDA staff who received an increased workload related to higher numbers of submissions for review, as noted above. FDA exceeded the 90 percent on-time performance goal for special protocol assessments. However, FDA performance, related to the remaining procedural and processing goals, fell short of the FY 2006 performance goal levels.
In 1992, Congress passed PDUFA, authorizing FDA to collect fees from companies that produce and submit applications for marketing human drug and biological products. The original PDUFA had a 5-year time limit that ended in 1997. This is the same year Congress passed the FDA Modernization Act (FDAMA), which contained a 5-year reauthorization of PDUFA (PDUFA II) that ended on September 30, 2002. When Congress passed the Public Health Security and Bioterrorism Preparedness and Response Act of 2002 (the Bioterrorism Act), it extended the PDUFA program for 5 more years (PDUFA III). PDUFA III is scheduled to come to an end on September 30, 2007. Information about PDUFA III, including the text of the amendments and the performance goals and procedures, can be found at: www.fda.gov/oc/pdufa.
PDUFA requires FDA to submit two annual reports to the President and the Congress for each fiscal year during which fees are collected: 1) a performance report due within 60 days of the end of the fiscal year, and 2) a financial report due within 120 days of the end of the fiscal year. This document addresses the first of these requirements for FY 2006. This year's report covers FDA's progress in meeting the quantifiable PDUFA review goals for FY 2005 and FY 2006 submissions and the FY 2006 procedural and processing goals. The report also describes FDA's progress in accomplishing new management initiatives and in meeting the information technology commitments of PDUFA III.
PDUFA provides FDA revenue to hire additional reviewers and support staff and upgrade its information technology systems to speed up the application review process for new drugs and biological products without compromising FDA's traditionally high standards for approval. Under PDUFA, FDA is committed to achieve certain performance goals that apply to the review of original and resubmitted new product applications and efficacy and manufacturing supplements to approved applications. FDA is also committed to achieve certain procedural and processing goals aimed at facilitating and assuring quality in new drug development.
During the first few years of PDUFA I, FDA eliminated backlogs of original applications and supplements that had formed in earlier years when the program had fewer resources. Over the course of PDUFA I, FDA agreed to review and act on a progressively increasing proportion of original NDAs, BLAs, and efficacy supplements within 12 months and resubmissions and manufacturing supplements within 6 months. FDA also agreed to review and act on 90 percent of priority NDAs, BLAs, and efficacy supplements (submissions that are for products providing significant therapeutic gains) submitted in FY 1997 within 6 months. Over the course of PDUFA I, FDA exceeded all of these performance goals.
In 1997, Congress passed FDAMA and reauthorized PDUFA (PDUFA II) for 5 more years. Under PDUFA II, most review times were shortened and FDA met or exceeded nearly all its review goals. PDUFA II expanded the scope of PDUFA work by including new goals intended to improve communication between FDA and application sponsors during the drug development process. These goals specified time frames for scheduling meetings, responding to various sponsor submissions, such as special protocols and responses to clinical holds, and other activities.
In 2002, Congress passed the Bioterrorism Act, which included an extension of PDUFA (PDUFA III) for 5 more years, FY 2003 through FY 2007. PDUFA III review performance goals and the procedural and processing goals are largely the same as the PDUFA II FY 2002 performance levels for these goals. PDUFA III establishes several new initiatives to improve application submissions and FDA-sponsor interactions during drug development and application review. In addition, it authorizes FDA to spend user fee funds on certain aspects of postmarket risk management, including surveillance of products approved after October 1, 2002, for up to 3 years.
PDUFA-enabled improvements in application quality and review efficiency have had an impact on the overall time to marketing approval. FDA tracks a variety of metrics related to the process of human drug review. The time-to-approval statistics are affected by a number of factors, including the total number of NDA and BLA submissions as well as the overall quality of submitted applications, the number of newly submitted priority applications, and the number of review staff relative to the review workload. These factors can vary from year to year; the charts that follow provide an update on trends in submissions and overall approval times.
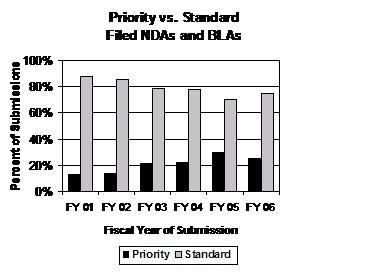
Total Number of NDA and BLA Applications Submitted in FY 2006 was the Second Highest Since FY 2000. Combined numbers of NDA and BLA priority and standard applications submitted appear to be returning to the higher levels seen under PDUFA II (see graph below). The number of NDA and BLA applications submitted and filed increased to pre-FY 2001 levels in two of the last 3 fiscal years, including FY 2006.
Priority Applications Filed Under PDUFA III Remain at High Levels. The number of priority applications, which represent significant therapeutic gains, steadily rose over the previous 5 years (FY 2001 to FY 2005) but leveled off in FY 2006. Priority NDA and BLA applications represent approximately one of every four NDA and BLA applications received by FDA (see graph below). The number of standard NDA and BLA applications submitted in FY 2006 increased after 5 straight years of decreases (FY 2001 to FY 2005).
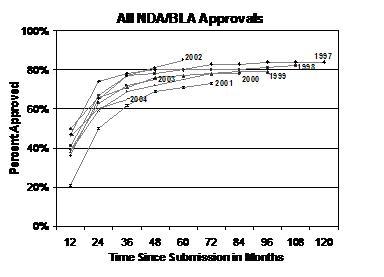
Historical Data Indicate that Approximately 80 Percent of Applications Submitted Reach Approval. A review of NDA and BLA approvals between FY 1997 and FY 2004 shows that most applications are approved within the first 2 years of submission to FDA. Within 24 months of submission, the percentage of approvals ranged from 51 percent in FY 2001 to 75 percent in FY 1997 with most cohorts between the 60 to 70 percent level. Based on historical trends, approximately 80 percent of NDA and BLA applications combined are approved within 5 years after submission (see graph below).
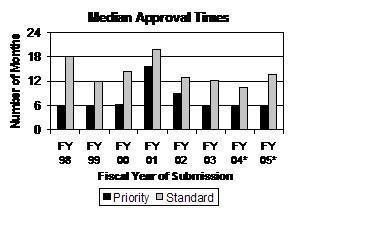
Median Time to Approval Remained Steady in FY 2005 for Priority Applications and Returned To Pre-FY 2004 Levels for Standard Applications. Estimated median time for approval of priority applications is 6.0 months for FY 2005. This is the third straight year (FY 2003 to FY 2005) for these historically low levels (see graph below). Based on applications approved through September 30, 2006, and historical data indicating close to 80 percent of all filed applications will eventually be approved (see graph above), the estimated median approval time for priority applications for FY 2004 and FY 2005 is 6.0 months. The estimated median approval time for standard applications in FY 2005 was 13.7 months, close to the median approval times for FY 2002 and FY 2003.
 D
D
*Estimated
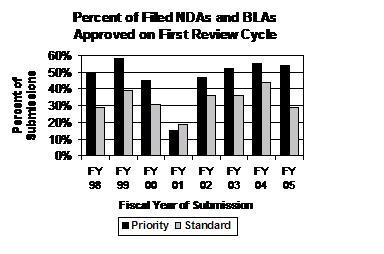
Percentage of First Cycle Approvals for Priority NDAs and BLAs Remained Above 50 Percent for the Third Straight Year. The percentage of priority NDA and BLA applications that were approved in the first review cycle from FY 2003 to FY 2005 was 52 percent, 55 percent, and 54 percent, respectively (see graph below). The percentage of standard applications approved in the first review cycle fell in FY 2005 to 29 percent.
Most Workload Categories Increased in FY 2006. FDA has seen significant variations to its workload under PDUFA III as defined by increasing numbers of product submissions and procedural and processing requests. No single year stands out with respect to across the board increases and decreases. In most categories, FY 2006 workload was higher than FY 2005. Concurrently, FDA reviewers faced significant increases in their workloads with respect to procedural and processing goals (see table below).
| Submission/Request | FY 2003 | FY 2004 | FY 2005 1 | FY 2006 |
|---|---|---|---|---|
| Original NDAs and BLAs Filed | 109 | 129 | 111 | 122 |
| Resubmitted NDAs and BLAs | 74 | 85 | 59 | 60 |
| NDA and BLA Efficacy Supplements | 153 | 204 | 158 | 182 |
| Resubmitted Efficacy Supplements | 59 | 58 | 48 | 37 |
| NDA and BLA Manufacturing Supplements | 2,598 | 2,500 | 2,532 | 2,679 |
| Meetings Scheduled | 2,002 | 2,125 | 2,230 | 2,266 |
| Special Protocol Assessments | 293 | 346 | 396 | 405 |
| Responses To Clinical Holds | 136 | 135 | 130 | 147 |
| Major Dispute Resolutions | 20 | 10 | 9 | 9 |
Review Performance At-A-Glance for FY 2005 and FY 2006
The tables below summarize FDA's review performance on the FY 2005 application submissions, and the preliminary performance in reviewing FY 2006 application submissions, and meeting other performance goals.
This section updates FDA's review performance on the FY 2005 application submissions and evaluates FDA's performance in reviewing FY 2006 application submissions and meeting other PDUFA performance goals. The following information refers to FDA performance presented in this section.
The table below summarizes the annual review time goals for original NDAs and BLAs. Over the 5-year period defined by PDUFA III, the goal of reviewing 90 percent of priority applications within 6 months and standard applications within 10 months remains constant.
| Original Application Type |
Review Time Goal | Performance Goal FY 2003 – FY 2007 Submissions |
|---|---|---|
| Priority | 6 months | 90% on time |
| Standard | 10 months |
Workload
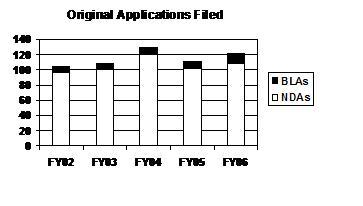
The total number of original applications in FY 2006 increased by 10 percent over the FY 2005 level (see graph and table below). In recent years, the workload has varied year to year primarily due to the number of standard applications. The number of priority applications increased 4 straight years before decreasing in FY 2006; the number of priority NMEs was at the lowest level in 5 years.
Original Applications Filed
(Priority / Standard)
| Type | FY 02 | FY 03 | FY 04 | FY 05 1 | FY 06 2 |
|---|---|---|---|---|---|
| NDAs | 96 (12/84) |
101 (19/82) |
120 (26/94) |
102 (29/73) |
109 (23/86) |
| BLAs | 9 (3/6) |
8 (4/4) |
9 (3/6) |
9 (6/3) |
13 (8/5) |
| PDUFA Total | 105 (15/90) |
109 (23/86) |
129 (29/100) |
111 (35/76) |
122 (31/91) |
| NMEs 3 | 22 (8/14) |
28 (12/16) |
30 (16/14) |
30 (15/15) |
22 (7/15) |
Performance
FY 2005 Submissions
FDA missed the 90 percent on-time review performance goal by 1 percent for priority NDAs and BLAs. The 90 percent on-time review performance goal was exceeded for priority NMEs and BLAs, and for all standard NDAs, NMEs, and BLAs in FY 2005. FDA reviewed and acted on most (31 of 35) priority applications within 6 months. FDA reviewed and acted on all but one (73 of 74) standard applications within 10 months (see table below). With two standard applications pending and not overdue as of September 30, 2006, FDA will exceed the on-time PDUFA review goal for standard applications.
| Original Application Type |
Review Within | Type | Reviewed and Acted On |
Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|---|
| Priority | 6 months |
All Applications |
35 | 31 | 89% | 90% |
| NMEs & BLAs | 21 | 20 | 95% | 90% | ||
| Standard | 10 months | All Applications |
74 | 73 | 99% | 90% |
| NMEs & BLAs | 17 | 16 | 94% | 90% |
FY 2006 Submissions
As of September 30, 2006, over half (19 of 31) of the priority applications filed in FY 2006 had been reviewed and acted on; and all but one (18 of 19) met the 6-month review performance goal. Approximately one-tenth (9 of 91) of the standard applications received had been reviewed and acted on; and all met the 10-month review performance goal (see table below). With submissions still pending and not overdue, it is too early to make a final performance determination for FY 2006.
| Original Application Type |
Review Within | Type | Reviewed and Acted On |
Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|---|
| Priority | 6 months |
All Applications |
19 | 18 | 95% | 90% |
| NMEs & BLAs | 10 | 9 | 90% | 90% | ||
| Standard | 10 months |
All Applications |
9 | 9 | 100% | 90% |
| NMEs & BLAs | 3 | 3 | 100% | 90% |
The table below summarizes the annual review time goals for resubmitted NDA and BLA applications. A resubmission is a firm's response after an FDA action of "approvable," "not approvable," or "complete response" on an application. The applicable performance goal for a resubmission is determined by the year in which the resubmission itself is received, rather than the year in which the original application was submitted. 4 Over the 5-year period defined by PDUFA III, the goal of reviewing 90 percent of Class 1 resubmitted applications within 2 months and Class 2 resubmitted applications within 6 months remains constant.
| Resubmitted Application Type | Review Time Goal | Performance Goal FY 2003 – FY 2007 Submissions |
|---|---|---|
| Class 1 | 2 months | 90% on time |
| Class 2 | 6 months | 90% on time |
Workload
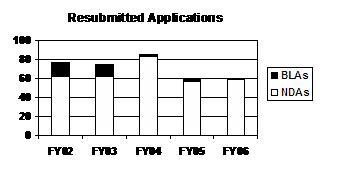
The total number of resubmitted applications was about the same for FY 2005 and FY 2006. The number of NDA resubmitted applications has been relatively level for 4 of the past 5 years; the sole exception was FY 2004. Resubmitted BLA applications have been at a relatively low level for 3 straight years with only one submitted in FY 2006 (see graph and table below).
Resubmitted Applications
(Class 1 / Class 2)
| Type | FY 02 | FY 03 | FY 04 | FY 05 1 | FY 06 |
|---|---|---|---|---|---|
| NDAs | 62 (20/42) |
62 (24/38) |
83 (21/62) |
56 (21/35) |
59 (20/39) |
| BLAs | 15 (2/13) |
12 (1/11) |
2 (1/1) |
3 (0/3) |
1 (0/1) |
| PDUFA Total | 77 (22/55) |
74 (25/49) |
85 (22/63) |
59 (21/38) |
60 (20/40) |
Performance
FY 2005 Resubmissions
The 90 percent on-time review performance goal was met for Class 1 and exceeded for Class 2 resubmissions in FY 2005. FDA reviewed and acted on all but two (19 of 21) Class 1 resubmitted applications within 2 months and reviewed and acted on all but three (35 of 38) Class 2 resubmitted application within 6 months (see table below).
Resubmitted Application Type |
Review Within | Reviewed and Acted On |
Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| Class 1 | 2 months | 21 | 19 | 90% | 90% |
| Class 2 | 6 months | 38 | 35 | 92% | 90% |
FY 2006 Resubmissions
As of September 30, 2006, most (18 of 20) of the Class 1 resubmissions received in
FY 2006 have been reviewed and acted on; all had met the 2-month review time goal (see table below). A little over half (22 of 40) of the Class 2 resubmissions received in
FY 2006 have been reviewed and acted on; all had met the 6-month review time goal. While FDA is assured of meeting the review time goal for Class 1 resubmissions, with Class 2 resubmissions still pending and not overdue, it is too early to make a final performance determination for FY 2006.
| Resubmitted Application Type |
Review Within | Reviewed and Acted On |
Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| Class 1 | 2 months | 18 | 18 | 100% | 90% |
| Class 2 | 6 months | 22 | 22 | 100% | 90% |
The table below summarizes the annual review time goals for original efficacy supplements to NDAs and BLAs. Over the 5-year period defined by PDUFA III, the goal of reviewing 90 percent of priority supplements within 6 months and standard supplements within 10 months remains constant.
| Efficacy Supplement Type | Review Time Goal | Performance Goal FY 2003 – FY 2007 Submissions |
|---|---|---|
| Priority | 6 months | 90% on time |
| Standard | 10 months | 90% on time |
Workload
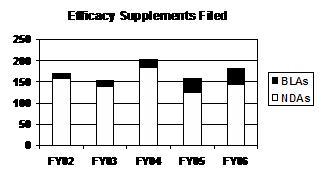
The total number of efficacy supplements received during the 5-year period (FY 2002 to FY 2006) has alternately decreased and increased. This fluctuation is a result of the number of NDA efficacy supplements, primarily standard, filed each year. The number of BLA efficacy supplements filed (mostly standard) has increased for 5 straight years (see graph and table below).
Efficacy Supplements Filed
(Priority / Standard)
| Type | FY 02 | FY 03 | FY 04 | FY 05 1 | FY 06 |
|---|---|---|---|---|---|
| NDAs | 159 (31/128) |
138 (35/103) |
183 (48/135) |
125 (34/91) |
145 (28/117) |
| BLAs | 11 (4/7) |
15 (2/13) |
21 (2/19) |
33 (7/26) |
37 (7/30) |
| PDUFA Total | 170 (35/135) |
153 (37/116) |
204 (50/154) |
158 (41/117) |
182 (35/147) |
Performance
FY 2005 Submissions
The 90 percent on-time review performance goal was exceeded for both priority and standard efficacy supplements in FY 2005. FDA reviewed and acted on all priority efficacy supplements within 6 months. FDA reviewed and acted on all but four (112 of 116) standard efficacy supplements within 10 months (see table below). With one standard efficacy supplement pending and not overdue as of September 30, 2006, FDA will exceed the on-time PDUFA review goal for standard efficacy supplements.
Efficacy Supplement Type |
Review Within | Reviewed and Acted On |
Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| Priority | 6 months | 41 | 41 | 100% | 90% |
| Standard | 10 months | 116 | 112 | 97% | 90% |
FY 2006 Submissions
As of September 30, 2006, almost two-thirds (22 of 35) of the priority efficacy supplements filed in FY 2006 had been reviewed and acted on; and all met the 6-month review performance goal. Almost one-fifth (29 of 147) of the standard efficacy supplements received had been reviewed and acted on; and all met the 10-month review performance goal (see table below). With submissions still pending and not overdue, it is too early to make a final performance determination for FY 2006.
| Efficacy Supplement Type |
Review Within | Reviewed and Acted On |
Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| Priority | 6 months | 22 | 22 | 100% | 90% |
| Standard | 10 months | 29 | 29 | 100% | 90% |
The table below summarizes the annual review time goals for resubmitted efficacy supplements to NDAs and BLAs. This is the fourth year for this goal under PDUFA III. For Class 1 resubmissions, the goal progresses from reviewing 30 percent of FY 2003 resubmissions in 2 months to 90 percent by FY 2007. Over the 5-year period defined by PDUFA III, the goal of reviewing 90 percent of Class 2 resubmissions within 6 months remains constant.
| Resubmitted Efficacy Supplement Type |
Review Time Goal |
Performance Goal | ||||
|---|---|---|---|---|---|---|
| FY 03 | FY 04 | FY 05 | FY 06 | FY 07 | ||
| Class 1 | 2 months | 30% | 50% | 70% | 80% | 90% |
| 4 months | -- | 90% | 90% | 90% | -- | |
| 6 months | 90% | -- | -- | -- | -- | |
| Class 2 | 6 months | 90% | 90% | 90% | 90% | 90% |
Workload
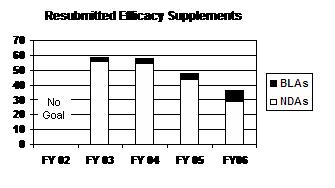
The total number of resubmitted efficacy supplements has decreased each year from FY 2003 to FY 2006. This is a result of fewer NDA efficacy supplement resubmissions each year. BLA efficacy supplement resubmissions have increased, including a doubling from four to eight from FY 2005 to FY 2006 (see graph and table below).
Resubmitted Efficacy Supplements
(Class 1 / Class 2)
| Type | FY 02 |
FY 03 |
FY 04 |
FY 051 |
FY 06 |
|---|---|---|---|---|---|
| NDAs | -- | 56 (16/40) |
55 (32/23) |
44 (23/21) |
29 (12/17) |
| BLAs | -- | 3 (1/2) |
3 (3/0) |
4 (1/3) |
8 (2/6) |
| PDUFA Total | -- | 59 (17/42) |
58 (35/23) |
48 (24/24) |
37 (14/23) |
Performance
FY 2005 Resubmissions
The on-time review performance goals were exceeded for both Class 1 and Class 2 efficacy supplement resubmissions in FY 2005. FDA reviewed and acted on all Class 1 resubmitted efficacy supplements within both the 2-month and 4-month review performance goals. FDA reviewed and acted on all but one (23 of 24) Class 2 resubmitted efficacy supplement within the 6-month review performance goal (see table below).
Resubmitted Efficacy Supplement Type |
Review Within | Reviewed and Acted On |
Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| Class 1 | 2 months | 24 | 24 | 100% | 70% |
| 4 months | 24 | 24 | 100% | 90% | |
| Class 2 | 6 months | 24 | 23 | 96% | 90% |
FY 2006 Resubmissions
As of September 30, 2006, most (9 of 14) of the Class 1 resubmitted efficacy supplements had been reviewed and acted on; and all met the 2-month and the 4-month review performance goals. Most (15 of 23) of the Class 2 resubmitted efficacy supplements had been reviewed and acted on; and all met the 6-month review performance goal (see table below). With resubmissions still pending and not overdue, it is too early to make a final performance determination for FY 2006.
| Resubmitted Efficacy Supplement Type |
Review Within | Reviewed and Acted On |
Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| Class 1 | 2 months | 9 | 9 | 100% | 80% |
| 4 months | 9 | 9 | 100% | 90% | |
| Class 2 | 6 months | 15 | 15 | 100% | 90% |
The table below summarizes the annual review time goals for manufacturing supplements to NDAs and BLAs. Over the 5-year period defined by PDUFA III, the performance goal for manufacturing supplements that require FDA's approval before the changes can be enacted is 90 percent of supplements within 4 months of submission. The PDUFA performance goal for manufacturing supplements that do not require FDA's approval before the changes can be enacted is 90 percent of supplements within 6 months of submission. The manufacturing supplement goals remain constant.
| Manufacturing Supplement Type |
Review Time Goal | Performance Goal FY 2003 – FY 2007 Submissions |
|---|---|---|
| Prior Approval Required | 4 months | 90% on time |
| Prior Approval Not Required | 6 months | 90% on time |
Workload
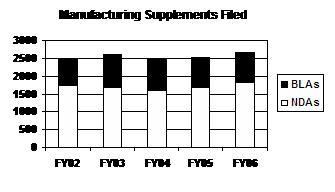
The total number of manufacturing supplements filed has risen over the past 3 years to a 5-year high in FY 2006. BLA supplements have represented about one-third of all manufacturing supplements from FY 2002 to FY 2006 (see graph and table below).
Manufacturing Supplements Filed
(Prior Approval / No Prior Approval)
| Type | FY 02 |
FY 031 |
FY 04 |
FY 051 |
FY 06 |
|---|---|---|---|---|---|
| NDAs | 1,759 (602/1,157) |
1,696 (617/1,079) |
1,617 (524/1,093) |
1,695 (630/1,065) |
1,824 (577/1,247) |
| BLAs | 717 (228/489) |
902 (303/599) |
883 (299/584) |
837 (257/580) |
855 (307/548) |
| PDUFA Total | 2,476 (830/1,646) |
2,598 (920/1,678) |
2,500 (823/1,677) |
2,532 (887/1,645) |
2,679 (884/1,795) |
Performance
FY 2005 Submissions
The 90 percent on-time review performance goal was exceeded for both types of manufacturing supplements in FY 2005. FDA reviewed and acted on almost all (869 of 887) manufacturing supplements that required prior approval within the 4-month review performance goal. FDA also reviewed and acted on almost all (1,625 of 1,640) manufacturing supplements not requiring prior approval within the 6-month review performance goal (see table below). With five manufacturing supplements not requiring prior approval, pending and overdue as of September 30, 2006, FDA will exceed the on-time PDUFA review goal.
Manufacturing |
Review Within |
Reviewed and |
Number on Time |
Percent on Time |
PDUFA |
Prior Approval |
4 months |
887 |
869 |
98% |
90% |
Prior Approval |
6 months |
1,640 |
1,625 |
99% |
90% |
FY 2006 Submissions
As of September 30, 2006, over two-thirds (617 of 884) of the manufacturing supplements requiring prior approval had been reviewed and acted on; and 96 percent (594 of 617) were reviewed within the 4-month review performance goal. Over one-half (965 of 1,795) of the manufacturing supplements not requiring prior approval had been reviewed and acted on; and 99 percent (959 of 965) were reviewed within the 6-month review performance goal (see table below). With submissions still pending and not overdue, it is too early to make a final performance determination for FY 2006.
| Manufacturing Supplement Type |
Review Within | Reviewed and Acted On |
Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| Prior Approval Required |
4 months | 617 | 594 | 96% | 90% |
| Prior Approval Not Required |
6 months | 965 | 959 | 99% | 90% |
This section presents FDA's performance in achieving the FY 2006 procedural and processing goals and accomplishments for PDUFA III initiatives and commitments. The following information refers to FDA performance presented in this section.
The procedural and processing goals FDA committed to achieve were designed to improve application submissions and FDA-sponsor interactions during new drug development and application review. The table below summarizes the meeting management goals that address meeting requests, scheduling meetings, and preparing meeting minutes.
| Action | Performance Goal | Performance Level FY 2003 – FY 2007 |
|---|---|---|
| Meeting Requests |
Notify requestor of formal meeting in writing within 14 days of request. | 90% on time |
| Scheduling Meetings | Schedule meetings within goal date (within 30 days of receipt of request for Type A meetings, 60 days for Type B meetings, and 75 days for Type C meetings). If the requested date for any of these types of meetings is greater than 30, 60, or 75 days, as appropriate, from the date the request is received by FDA, the meeting date should be within 14 days of the requested date. | 90% on time |
| Meeting Minutes |
FDA-prepared minutes, clearly outlining agreements, disagreements, issues for further discussion, and action items will be available to the sponsor within 30 days of meeting. | 90% on time |
Workload
The number of meeting requests and, subsequently, the number of meetings scheduled increased for the fourth straight year in FY 2006 (see graph and table below).
Meeting Management
| Type | FY 02 |
FY 03 |
FY 04 |
FY 051 |
FY 06 |
|---|---|---|---|---|---|
| Meeting Request Notifications | 1,745 | 2,119 | 2,284 | 2,487 | 2,548 |
| Scheduling Meetings | 1,643 | 2,002 | 2,125 | 2,230 | 2,266 |
| Meeting Minutes | 1,503 | 1,761 | 1,854 | 1,901 | 1,870 |
FY 2006 Performance
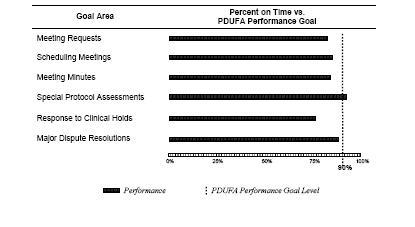
As of September 30, 2006, FDA had responded to virtually all (2,537 of the 2,548) of the meeting requests, most (2,203 of 2,266) meetings granted had been scheduled, and over three-fourths (1,490 of 1,870) of meetings held had minutes issued. Preliminary performance indicated FDA was not meeting the 90 percent on-time performance goals for meeting management. While activities are still pending and not overdue, completing these activities on-time will not raise overall performance enough to meet the performance goals (see table below).
| Total | Met Goal | Missed Goal 5 | Pending Within Goal | Percent on Time 6 | PDUFA Performance Goal |
|||
|---|---|---|---|---|---|---|---|---|
| Meeting Requests | CBER | 298 | 290 | 6 | 2 |
|
|
|
| CDER | 2,250 | 1,778 | 463 | 9 | ||||
| Combined | 2,548 | 2,068 | 469 | 11 | 82% | 90% | ||
| Scheduling Meetings 7 | Type A | CBER | 18 | 15 | 1 | 2 |
|
|
| CDER | 175 | 145 | 25 | 5 | ||||
| Type B | CBER | 167 | 137 | 7 | 23 | |||
| CDER | 1,215 | 968 | 229 | 18 | ||||
| Type C | CBER | 79 | 70 | 2 | 7 | |||
| CDER | 612 | 534 | 70 | 8 | ||||
| All | CBER | 264 | 222 | 10 | 32 | |||
| CDER | 2,002 | 1,647 | 324 | 31 | ||||
| Combined | 2,266 | 1,869 | 334 | 63 | 85% | 90% | ||
| Meeting Minutes |
CBER | 193 | 174 | 4 | 15 |
|
|
|
| CDER | 1,677 | 1,075 | 237 | 365 | ||||
| Combined | 1,870 | 1,249 | 241 | 380 | 84% | 90% | ||
The table below summarizes the annual performance goal for the response to the requests for special protocol assessments. Over the 5-year period defined by PDUFA III, the goal of responding to 90 percent of sponsors' requests for evaluation of protocol design within 45 days of receipt remains constant.
| Action | Performance Goal | Performance Level FY 2003 – FY 2007 |
|---|---|---|
| Special Protocol Question Assessment and Agreement | Respond to sponsor's request for evaluation of protocol design within 45 days of receipt. | 90% on time |
Workload
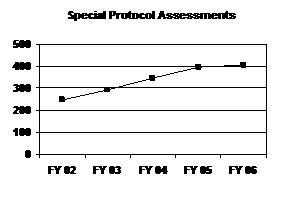
Special protocol assessment requests have increased for 5 straight years, although at a slower rate from FY 2005 to FY 2006 (see graph and table below).
Special Protocol Assessments
FY 02 |
FY 03 |
FY 04 |
FY 05 1 |
FY 06 |
|---|---|---|---|---|
| 248 | 293 | 346 | 396 | 405 |
FY 2006 Performance
As of September 30, 2006, FDA responded to most (373 of 405) of the sponsors' requests for evaluation of protocol designs received in FY 2006. FDA is exceeding the performance goal for response time to requests for special protocol assessments by responding to 92 percent (344 of 373) special protocol assessments on time. With submissions still pending and not overdue, it is too early to make a final performance determination for FY 2006.
Special Protocol Assessments (CBER / CDER)
| Total | Met Goal | Missed Goal | Pending Within Goal | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| 405 15/390) | 344 (12/332) | 29 (0/29) | 32 (3/29) | 92% | 90% |
The table below summarizes the annual performance goal for the response to clinical holds. Over the 5-year period defined by PDUFA III, the goal of responding to sponsor's complete response to a clinical hold within 30 days of receipt remains constant.
| Action | Performance Goal | Performance Level FY 2003 – FY 2007 |
|---|---|---|
| Response to Clinical Hold | Respond to sponsor's complete response to a clinical hold within 30 days of receipt. | 90% on time |
Workload
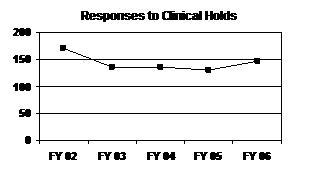
After decreasing the previous 3 years (FY 2003 to FY 2005), the number of responses to clinical holds in FY 2006 increased to the highest level since FY 2002 (see graph and table below).
Responses to Clinical Holds
| FY 02 | FY 03 | FY 04 | FY 051 | FY 06 |
|---|---|---|---|---|
| 171 | 136 | 135 | 130 | 147 |
FY 2006 Performance
As of September 30, 2006, FDA responded to most (133 of 147) of sponsors' complete responses to clinical holds received in FY 2006. However, FDA did not meet the on-time performance goal for responses to clinical holds. The preliminary data show that 76 percent were responded to within goal. There are 14 responses to clinical holds pending within goal; however, meeting the performance goal for all the remaining clinical hold responses will not enable FDA to meet the FY 2006 performance goal.
Responses to Clinical Holds (CBER / CDER)
| Total | Met Goal | Missed Goal | Pending Within Goal | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| 147 (43/104) | 101 (37/64) | 32 (2/30) | 14 (4/10) | 76% | 90% |
The table below summarizes the annual performance goal for the response to major dispute resolutions. Over the 5-year period defined by PDUFA III, the goal of responding to sponsor's appeal of decision within 30 days of receipt remains constant.
| Action | Performance Goal | Performance Level FY 2003 – FY 2007 |
|---|---|---|
| Major Dispute Resolution | Respond to sponsor's appeal of decision within 30 days of receipt. | 90% on time |
Workload
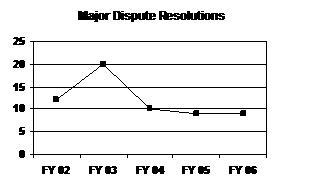
FDA has been addressing close to 10 major dispute resolutions each year, with the exception of FY 2003, when 20 major dispute resolutions were addressed (see graph and table below).
Major Dispute Resolutions
| FY 02 | FY 03 | FY 04 | FY 05 | FY 06 |
|---|---|---|---|---|
| 12 | 20 | 10 | 9 | 9 |
FY 2006 Performance
As of September 30, 2006, FDA responded to most (8 of 9) sponsors' appeals of decisions received in FY 2006. There is one appeal still pending within goal; however, the eventual response to this appeal will not enable FDA to meet the FY 2006 performance goal.
Major Dispute Resolutions (CBER / CDER)
| Total | Met Goal | Missed Goal | Pending Within Goal | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| 9 (0/9) | 7 (0/7) | 1 (0/1) | 1 (0/1) | 88% | 90% |
Goal - Report Substantive Deficiencies (or Lack of Same) Within 14 Days After the 60-Day Filing Date for Original BLAs, NDAs, and Efficacy Supplements
The table below summarizes the annual review time goals for first cycle filing review notifications for original NDAs, BLAs, and efficacy supplements. This is the fourth year for this goal under PDUFA III. FDA is to report substantive deficiencies (or lack of same) identified during the initial filing review to the sponsor by letter, telephone conference, facsimile, secure e-mail, or other expedient means within 14 days after the 60-day filing date. Performance levels progress from 50 percent on time for FY 2003 submissions to 90 percent for FY 2005 to FY 2007 submissions.
First Cycle Filing Review Notification Type |
Review Time Goal | Performance Level | ||||
|---|---|---|---|---|---|---|
| FY 03 | FY 04 | FY 05 | FY 06 | FY 07 | ||
| Original NDAs and BLAs | Within 14 days after 60-day filing date |
50% | 70% | 90% | 90% | 90% |
Workload
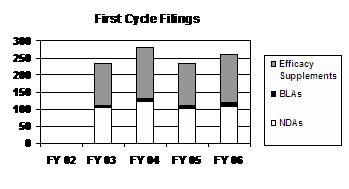
The total number of first cycle filings has fluctuated over the past 4 years, with FY 2006 representing an increase of 10 percent over the FY 2005 level. The number of first cycle filings for NDAs, BLAs, and efficacy supplements increased in FY 2006 (see graph and table below).
First Cycle Filings
| Type | FY 02 | FY 03 | FY 04 | FY 05 1 | FY 06 |
|---|---|---|---|---|---|
| NDAs | -- | 104 | 123 | 102 | 108 |
| BLAs | -- | 8 | 9 | 9 | 13 |
| Total | -- | 112 | 132 | 111 | 121 |
| Efficacy Supplements 8 | -- | 121 | 147 | 124 | 140 |
Performance
FY 2005 Submissions
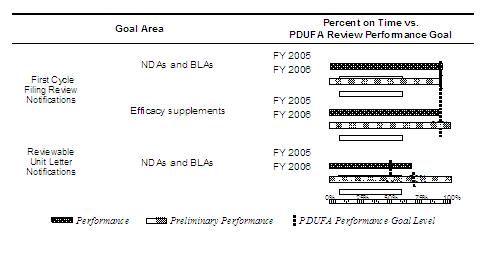
The on-time review performance goals were exceeded for NDA and BLA first cycle filing review notifications in FY 2005. FDA completed initial filing reviews for most (102 of 111) original NDAs and BLAs within 14 days after the 60-day filing date. FDA completed initial filing reviews for most (110 of 124) efficacy supplements within 14 days after the 60-day filing date but missed the performance goal by 1 percent (see table below).
| First Cycle Filing Review Notification Type |
Review Within | Initial Filing Reviews | Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| NDAs and BLAs | Within 14 days after 60-day filing date |
111 | 102 | 92% | 90% |
| Efficacy Supplements | Within 14 days after 60-day filing date |
124 | 110 | 89% | 90% |
FY 2006 Submissions
As of September 30, 2006, over four-fifths (100 of 121) of NDAs and BLAs had received an initial filing review; and 90 percent (90 of 100) were reviewed within 14 days after the 60-day filing date. Over four-fifths (117 of 140) of efficacy supplements were reviewed, with 94 percent (110 of 117) reviewed within goal (see table below). With submissions still pending and not overdue, it is too early to make a final performance determination for FY 2006.
| First Cycle Filing Review Notification Type |
Review Within | Initial Filing Reviews | Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| NDAs and BLAs | Within 14 days after 60-day filing date |
100 | 90 | 90% | 90% |
| Efficacy Supplements | Within 14 days after 60-day filing date |
117 | 110 | 94% | 90% |
Goal – Issue Discipline Review Letters for Pre-submitted "Reviewable Units" of NDAs and BLAs
The table below summarizes the annual review time goals for reviewable unit letter notifications for NDAs and BLAs. This is the third year for this goal under PDUFA III. Under the Continuous Marketing Application Pilot 1 program, applicants may submit a portion of their marketing application, reviewable unit (RU), before submitting the complete application for Fast Track Original NDAs and BLAs, based on meeting specific criteria for inclusion in the Pilot. An NDA or BLA may have more than one RU. Each RU is tracked independently. Under this goal, FDA is to issue discipline review letters for pre-submitted RUs to NDAs and BLAs within 6 months of receipt. Performance levels progress from 30 percent on time for FY 2004 submissions to 90 percent for FY 2007 submissions.
| Reviewable Unit Type | Review Time Goal | Performance Level | ||||
|---|---|---|---|---|---|---|
| FY 03 | FY 04 | FY 05 | FY 06 | FY 07 | ||
| NDA | 6 months | -- | 30% | 50% | 70% | 90% |
| BLA | 6 months | |||||
Workload
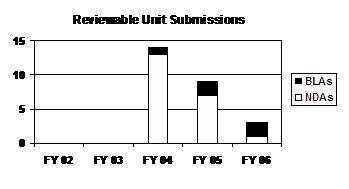
The total number of NDA reviewable unit submissions decreased for 3 straight years (see graph and table below).
Reviewable Unit Submissions
| Type | FY 02 | FY 03 | FY 04 | FY 05 | FY 06 |
|---|---|---|---|---|---|
| NDAs | -- | -- | 13 | 7 | 1 |
| BLAs | -- | -- | 1 | 2 | 2 |
| PDUFA Total | -- | -- | 14 | 9 | 3 |
Performance
FY 2005 Submissions
FDA performance on all reviewable unit letter notifications exceeded the 50 percent on-time review performance goal in FY 2005. FDA reviewed and acted all but three (6 of 9) reviewable unit submissions within 6 months (see table below).
| Reviewable Unit Type |
Review Within | Reviewed and Acted On |
Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| NDAs and BLAs | 6 months | 9 | 6 | 67% | 50% |
FY 2006 Submissions
As of September 30, 2006, two of the three NDA and BLA reviewable unit submissions had been reviewed and acted on; and both were reviewed within the 6-month review time goal (see table below). With one reviewable unit submission still pending and not overdue, it is too early to make a final performance determination for FY 2006.
| Reviewable Unit Type |
Review Within | Reviewed and Acted On |
Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| NDAs and BLAs | 6 months | 2 | 2 | 100% | 70% |
The management initiatives FDA committed to achieve under PDUFA III were designed to improve the overall application review process.
The first Continuous Marketing Application (CMA) pilot (Pilot 1) applies to fast track products that have demonstrated significant promise as a therapeutic advance in clinical trials, and will provide an early discipline review of the reviewable units (RUs) of the sponsor's NDA/BLA submitted in advance of the complete application. (The CMA Pilot 1 program became effective when the final guidance was published on October 6, 2003, and is available at: http://www.fda.gov/cder/guidance/5739-fnl.pdf.)
The second CMA pilot (Pilot 2) also applies to fast track products and provides for FDA-sponsor agreement to engage in frequent scientific feedback and interactions during the clinical trial phase of product development. (The CMA Pilot 2 program became effective when the final guidance was published on October 6, 2003, and is available at: http://www.fda.gov/cder/guidance/5740-fnl.pdf.)
FY 2006 Accomplishments: As of September 30, 2006, a cumulative total of 14 products had been identified for inclusion in the Pilot 1 program. Three RUs were received during FY 2006. As of September 30, 2006, two of the three RUs received had been reviewed and acted on; and both were within the goal time. Additionally, a total of 9 products were involved in the Pilot 2 program as of September 30, 2006.
In August 2005, FDA awarded a task order under an existing contract to evaluate the CMA Pilots. The evaluation was completed and the final report posted on the FDA PDUFA website, http://www.fda.gov/ope/CMA/CMAFinalReport.pdf, on June 22, 2006. There was no conclusive finding that indicates whether the Pilot 1 program should continue or be terminated. Findings showed that there were two different Pilot 2 approaches sponsors used to schedule exchanges with FDA—one approach established an estimated schedule in advance (the Fixed Schedule) and the other focused on when FDA could provide feedback based on the type of interaction (the Trigger Method), scheduling interactions as needed. It is too early in the Pilot 2 program to determine the value of its impact.
Approvals that take more than one review cycle to complete are generally not in the best interest of the public, the FDA, or the sponsor submitting the product application. Although additional review cycles are sometimes necessary to resolve important issues regarding safety, quality, or efficacy; in most cases, the extra cycles could be avoided, saving time and effort. For applications that are ultimately approved, the causes of multiple review cycles can include deficiencies in sponsors' applications, communication problems during the review process, or difficulty achieving final resolution on such topics as labeling.
Efforts to improve the first cycle review process include an initiative for notification of substantive deficiencies identified during the initial filing review for original NDAs and BLAs and an initiative to develop and publish Good Review Management Principles (GRMP) with provisions for both FDA reviewers and industry sponsors. The notification initiative was implemented on October 2, 2002. The final GRMP guidance was published on March 31, 2005, and is available at: http://www.fda.gov/cder/guidance/5812fnl.htm.
FY 2006 Accomplishments: As of September 30, 2006, 90 percent (90 of 100) of NDAs and BLAs and 94 percent (110 of 117) of efficacy supplements had received an initial filing review within the goal time.
In January 2005, FDA awarded a task order under an existing contract to conduct a retrospective analysis of first cycle reviews. The retrospective analysis was completed and the final report posted on the FDA PDUFA website, http://www.fda.gov/ope/pdufa/PDUFA1stCycle/pdufa1stcycle.pdf, on June 22, 2006. In December 2005, FDA awarded a task order under an existing contract to conduct a prospective analysis of first cycle reviews. Findings showed that: Priority and Fast-Track products have higher first-cycle approval rates; most products that fail to receive first-cycle approval have key deficiencies in only one or two categories, with an even breakdown between the categories of safety, efficacy, and chemistry; and effective communication and responsiveness to FDA inquiries marked first-cycle approvals while persisting disagreements over issue resolution were associated with approval delays.
Under the PDUFA III performance management goal, FDA will conduct initiatives that are targeted to improve the new drug review process. FDA will also contract with outside expert consultants for analysis, training, and technical assistance to help implement a quality systems approach to the new drug review process. In November 2004, FDA established a Quality Systems Group to coordinate the implementation of a quality management system for new drug review and PDUFA III performance management initiatives.
FY 2006 Accomplishments: Contracts were awarded in FY 2006 for such projects as: 1) a study of postmarketing commitments, 2) development of a quality management system for chemistry manufacturing and controls (CMCs) reviews, 3) development of a quality management system for CBER and CDER laboratories product performance data, 4) focus groups for physicians and pharmacists regarding drug safety, and 5) regulatory database training for reviewers.
Work continued on existing contracts for process improvements in CDER's Office of New Drugs and Office of Surveillance and Epidemiology, quality meeting minutes, leadership development training, and managerial costing.
Independent Consultants
This PDUFA III initiative allows a sponsor to request that FDA engage an independent expert consultant during the development period for certain biotechnology products. The consultant would be selected by FDA to assist in FDA's review of the protocol for the clinical studies that would support the claims for the product. This initiative is intended to facilitate product development. Final guidance was published on August 18, 2004, and is available at: http://www.fda.gov/cber/gdlns/bioclin.htm.
FY 2006 Accomplishments: No sponsors have requested assistance under the program.
The initiative to address postmarket risk before an application is submitted, during the review process, and during the peri-approval period (2 or 3 years post-approval), will facilitate postmarket risk management by helping FDA better understand any risks and by providing feedback to the sponsors. The following Guidance for Industry was published in the Federal Register on March 29, 2005:
FY 2006 Accomplishments: CBER/Division of Epidemiology (DE) reviewed three Pharmacovigilance Plans and three other postmarketing study plans. The Division also participated in six pre-BLA review meetings and four peri-approval meetings, all for PDUFA III products. CBER/DE also reviewed two postmarketing study plans and evaluated one active Phase IV study for a non-PDUFA III product that resulted in labeling changes.
The electronic applications and submissions commitments under PDUFA III were designed to improve the overall application review process.
Centralize the accountability and funding for all PDUFA Information Technology (IT) initiatives/activities under the leadership of the FDA Chief Information Officer (CIO).
FY 2006 Accomplishments: The accountability and funding for all PDUFA IT initiatives/activities were centralized under the leadership of the FDA CIO in FY 2003. In February 2006, FDA further strengthened the IT oversight to ensure business driven, enterprise-wide direction and management through the formation of the FDA Bioinformatics Board and the PDUFA Budget Review Board. The Bioinformatics Board coordinates and oversees all activities related to business automation planning, acquisition, and implementation decisions throughout FDA. FDA's approach is based on the premise that oversight of the design, building, and maintenance of such an infrastructure must be both business-driven and business-owned. In support of this FDA approach and to ensure that the PDUFA Program needs are met, FDA established the PDUFA Budget Review Board to oversee all PDUFA-related spending. The Bioinformatics Board reports to FDA Management Council and the CIO is a member of all three of these oversight organizations. The CIO ensures that FDA IT investments are in alignment with business requirements by providing leadership and oversight for the design, development, implementation, and support of all IT initiatives/activities.
Periodically review and evaluate the progress of IT initiatives against project milestones. This includes, on an annual basis, an assessment of progress against PDUFA III IT goals and established program milestones, including appropriate changes to plans.
FY 2006 Accomplishments: This FY 2006 PDUFA Performance Report to the President and the Congress satisfies the annual requirement. In addition, FDA reported IT progress to stakeholders at the PDUFA IT quarterly briefings and through PhRMA/BIO PDUFA updates.
Implement a common solution for the secure exchange of application content.
FY 2006 Accomplishments: FDA has continued to participate and provide guidance on the Signatures and Authentication for Everyone (SAFE) standard for the biopharmaceutical industry. During FY 2006, FDA worked with the SAFE team to develop a SAFE-FDA Auditor Familiarization Program to provide industry and FDA auditors the background, insight, and tools for the auditing/inspection process.
Deliver a single point of entry for the receipt and processing of all electronic submissions in a highly secure environment.
FY 2006 Accomplishments: In May 2006, the FDA Electronic Submissions Gateway (ESG) went into production. FDA ESG is an FDA-wide solution that enables the secure submission of electronic regulatory submissions. It is the central transmission or single point of entry for sending PDUFA regulatory submissions electronically to FDA. The electronic submission process encompasses the receipt, acknowledgment of receipt (to the sender), routing, and notification (to a receiving Center or Office) of the delivery of an electronic submission.
By the end of FY 2006, the ESG had received and processed over 33,000 premarketing and postmarketing submissions. Information on the ESG process and requirements is available at: http://www.fda.gov/esg/.
Provide a format and review system for the electronic submission of the Common Technical Document (e-CTD).
FY 2006 Accomplishments: In FY 2006, FDA enhanced the e-CTD review system to provide reviewers with additional search capabilities and to track the progress of the e-CTD submission review at the section level.
In FY 2006, there was a dramatic increase in the number of e-CTD submissions with approximately 4,000 e-CTD submissions received. Since FY2003, CBER and CDER have received over 5,000 e-CTD submissions. The e-CTD guidance and specifications are available at: http://www.fda.gov/cder/regulatory/ersr/ectd.htm.
Conduct an objective analysis and develop a plan for consolidation of PDUFA III IT infrastructure.
FY 2006 Accomplishments: In FY 2006 the IT Infrastructure Transformation (ITX) Program was established to lead FDA toward a unified and consistent approach in managing FDA's IT assets. The ITX Program encompasses the development of a unified strategy, roadmap, and implementation for infrastructure component areas that include: server and storage consolidation, application co-location, enterprise management capability, pre-production environment, asset management, disaster recovery, email consolidation (HHSMail), White Oak Data Center, and capacity management. The goals of the program are to increase end user computing reliability and availability and reduce IT operations and maintenance costs. The project phases will be staggered from FY 2006 to FY 2009, with the analysis, strategic approach, and roadmap for the projects defined in FY 2006 and 2007. Implementation is scheduled to take place in FY 2008 and FY 2009. The ITX Program is being performed in parallel with plans for the relocation of FDA Headquarters to the White Oak campus.
The following tasks were completed during FY 2006, as FDA moves towards consolidation of staff and IT infrastructure.
Implement Capability Maturity Model (CMM) and include other industry best practices to ensure quality, efficiency, and cost effectiveness.
FY 2006 Accomplishments: FDA continues to strengthen and improve IT project management capabilities to ensure that all IT projects follow standardized industry best practices. FDA has established project Stage Gate Review guidelines, conducted stage gate reviews, conducted post-implementation lessons-learned sessions for each major IT investment, and requires earned value management reporting on all IT investments. In addition, FDA continues to provide project management certification training.
Office of Information Technology (OIT)-CBER received certification for CMM Level 2 rating for its project management group in December 2005 and is now adopting the FDA Investment Life Cycle (ILC) procedures, which involve the use of standardized templates and stage gates.
OIT-CDER implemented the FDA ILC, with all new development and operational releases following standard procedures for communication, risk management, requirements management, and reporting. All release efforts are reviewed at each stage gate for adherence to the FDA ILC, projects goals, and financial performance.
For the ITX Program, a separate Project Management Office (PMO) was established. The ITX PMO coordinates with the ITX Integration Program Manager to ensure that the various ITX projects follow the FDA Software Development Life-Cycle and conduct stage gate reviews for each project.
Use the same software applications where common business needs exist.
FY 2006 Accomplishments: FDA implemented the Electronic Labeling Information Processing System (ELIPS). At the beginning of FY 2006, the ELIPS was implemented to handle the Electronic Labeling Rule requiring the submission of the content of labeling in electronic format for marketing applications. In the fourth quarter of FY 2006, the ELIPS was upgraded to handle the Requirements on Content and Format of Labeling for Human Prescription Drug and Biological Products (Physicians Labeling Rule), which amended the content and format of prescribing information for human drug and biologic products. The Physicians Labeling Rule requires that the prescribing information of new and recently approved products includes highlights of the prescribing information and a table of contents for the full prescribing information. Although the initial production releases of the ELIPS have been in CDER, the system will be expanded to CBER and other FDA Centers in future releases.
Additional information is available at: http://www.fda.gov/oc/datacouncil/spl.html and http://www.fda.gov/cder/regulatory/physLabel/default.htm.
Develop a PDUFA III IT 5-year plan.
An update to the March 2003 PDUFA IT Plan, that met the requirements of this performance goal, was completed in June 2004 and released at the September 2004 PDUFA IT quarterly briefing.
1. Numbers have been revised to reflect updated information not available for the FY 2005 PDUFA Performance Report.
2. The count of FY 2006 submissions assumes that all submissions received in the last 2 months of FY 2006 are filed. When FDA files a submission, it is deemed "complete" by PDUFA definition. FDA makes a filing decision within 60 days of an original application's receipt. All PDUFA review times are calculated from the original receipt date of the filed application.
3. FDA often receives multiple submissions for the same NME, which are all initially designated as NMEs. When FDA approves the first of the multiple submissions, the others are redesignated as non-NMEs.
4. Class 1 resubmissions are applications resubmitted after a complete response letter (or a not approvable or approvable letter) that include items listed on page A-7 in Appendix A. Class 2 resubmissions are applications resubmitted that include other items, such as those presented to an advisory committee.
5. Includes those with late actions and those still pending where the goal date has passed and which have not had actions.
6. Calculation based only on actions identified as being met or missed. Actions pending within goal were excluded from the calculation.
7. Not all meeting requests are granted.
8. The First Cycle Filing Review Notification goal applies to original NDAs, BLAs, and efficacy supplements only. It does not apply to NDA labeling supplements that contain clinical data, even though these are counted as efficacy supplements for other PDUFA performance purposes. Therefore, the number of filing review notifications for efficacy supplements is less than the total number of efficacy supplements filed (as shown on page 14).
 |
Department of Health and Human Services Food and Drug Administration |
 |
This report was prepared by FDA's Office of Planning in collaboration with the Center for Biologics Evaluation and Research (CBER) and the Center for Drug Evaluation and Research (CDER). For information on obtaining additional copies contact:
Office of Planning (HFP-10)
Food and Drug Administration
5600 Fishers Lane
Rockville, Maryland 20857
Phone: 301-827-5292
FAX: 301-827-5260
This report is available on the FDA Home Page at www.fda.gov/ope/pdufa/report2006/
![]()