


FDA UPDATE
Melissa Greenwald, M.D.
Division of Human Tissues
![]()
OCTGT, CBER, FDA
AATB 31st Annual Meeting
18 September 2007
Boston, MA
FDA Update
- Registration and Listing
- Exemptions and Alternatives
- Adverse Reaction (AR) Reporting
- HCT/P Deviations
- Publications
Registration
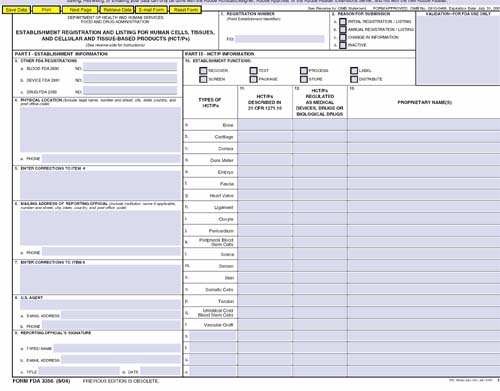
Registration
- Annual registration update required in December
- Electronically registered establishments were notified via email if not updated
- Electronic registration is encouraged—63% did so last year
Registration

- There is an expiration date in the top right corner of the form
- This is an expiration date of the form itself—we will continue to use this form until another form is approved and posted
- This is not an expiration date for your registration
- All registrations “expire” in December each year
Registration
- Changes to listing are required within 6 months
- Change of ownership or location required within 5 days
- Inactivation—check box #2(d)

Registration

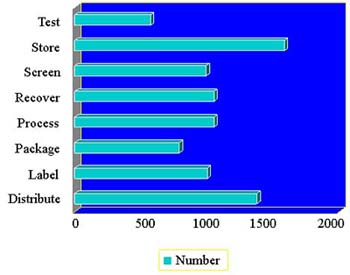
- There are 572 facilities registered with “test” as an establishment function (last year 612)
- “Test”and “Screen”refer to the donor, not the HCT/P
- You should list this function only if you perform donor testing, not if you send specimens out for testing
- Microbiologic testing of the HCT/P is considered processing
Failure to Register
- If you do not renew your annual registration CBER’sOffice of Compliance will contact district offices to follow-up
- A failure to register notice has been posted on CBER’s website www.fda.gov/cber/tissue/failreg.htm
- Information: www.fda.gov/cber/tiss.htm
- Electronic Form Instructions: http://www.fda.gov/cber/tissue/tisreginstr.htm
- Questions: tissuereg@fda.hhs.gov
Registration and Listing
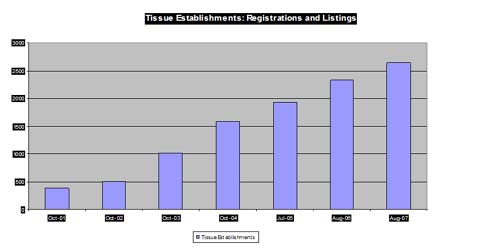
| Total (as of 8/3/07) = 2650 (531 inactive) Musculoskeletal/ocular = 1286 Hematopoietic stem cell = 654 Reproductive = 685 International = 186 (mostly stem cell) |
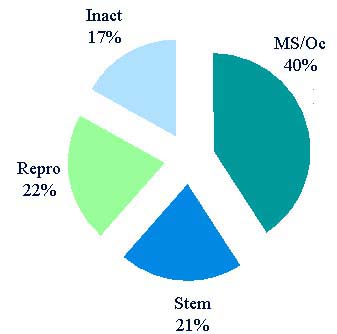 |
Note: Number of establishments by tissue type is not additive to the total beacuse of overlap.
Registration
 |
|
Proposed Rule for Registration and Listing for Foreign and Domestic Establishments
- Published August 29, 2006
- Comment period extended/ended 1/26/07
- Public meeting 12/11/06 discussed proposed changes to National Drug Code (NDC) system
- Would affect HCT/Ps licensed as biological products (e.g., hematopoietic stem cells and other cell therapy products)
- NDC # assigned by FDA
- Must be on the product label
Proposed Registration & Listing Rule Changes to Part 1271
- New definitions: importer, US agent
- Would require electronic registration unless a waiver is granted
- Foreign establishments importing HCT/Ps would submit information on all known US consignees
- Changes to ownership or location would be reported within 30 days rather than 5 days
Exemptions and Alternatives
- May request exemption or alternative from donor eligibility and CGTP requirements (1271.155)—currently does not include registration & listing
- Must be accompanied by supporting documentation justifying the request
- OCTGT SOPP 9151 6/13/06
- Division of Human Tissues coordinates
- Consults experts outside of division
- Draft letter presented to Tissue Policy Team for discussion/concurrence
- Center Director sign-off
Adverse Reactions
- Adverse reaction means a noxious and unintended response to any HCT/P for which there is a reasonable possibility that the HCT/P caused the response
- If adverse reaction is reportable, manufacturers must submit a MedWatch 3500A to FDA within 15 days of receipt of information
- Must also submit follow-up MedWatch report within 15 days of receipt of new information from the investigation of reportable adverse reactions (1271.3(y))
Adverse Reaction Reporting
- Manufacturers must investigate:
- Any adverse reaction involving a communicable disease related to an HCT/P that they made available for distribution.
- Manufacturers must report to FDA
- An adverse reaction involving a communicable disease if it:
- Is fatal
- Is life-threatening
- Results in permanent impairment of function or permanent damage to body structure; or
- Necessitates medical or surgical intervention, including hospitalization (1271.350)
How are Adverse Reactions Reported to FDA?
- For Manufacturers:
- Use Form FDA 3500A (MedWatch)
- Report w/in 15 days of receipt of information
- For Voluntary reporters:
- Use Form FDA 3500 (MedWatch)
- Recommend prompt reporting to HCT/P establishments
- FDA MedWatch reporting system http://www.fda.gov/medwatch
Adverse Reactions

- www.fda.gov/cber/gdlns/advhctp.htm
- Provides additional information and instructions for filling out the Medwatch Forms
Adverse Reaction Reports Internal Procedures
- SOPP 8508 describes procedures for handling AR reports—coordinating 5 offices within CBER www.fda.gov/cber/regsopp/8508.htm
- MedWatch reports involving “361”and biological product HCT/Ps come to CBER
- Follow-up on infectious adverse reactions
- Interoffice review; discussed at Tissue Safety
- Team meetings
- Seek more information from reporter and manufacturer as needed
Reported Adverse Reactions
| Tissue Type | CY 2006 | 2007 Total (through 8/3) |
|---|---|---|
| Blood Vessel | 3 (2%) | 1 (1.7%) |
| Bone | 38 (25.9%) | 11 (18.6%) |
| Cardiac | 6 (4.1%) | 1 (1.7%) |
| Ocular | 36 (24.5%) | 8 (13.6%) |
| MSK Soft | 30 (20.4%) | 19 (32.2%) |
| Skin | 29 (19.7%) | 16 (27.1%) |
| Tissue, NOS | 2 (1.4%) | 3 (5.1%) |
| Unassigned | 3 (2.0%) | 0 |
| Grand Total | 147 | 60 |
Reported Adverse Reactions By Outcomes
Calendar Year 2006 Totals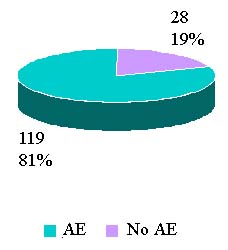 |
CY 2007 to date (Jan-3 Aug 2007)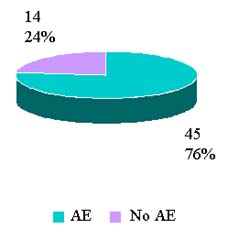 |
No adverse event—product problem, recall, or combination
Reported Adverse Events By Reporter
CY 2006 Reports
| Report Source | Number | Percent |
|---|---|---|
| Mfr. | 87 | 46.3 |
| Direct | 49 | 26.1 |
| MedSun | 17 | 9.0 |
| Others | 3 | 1.6 |
| Blank | 32 | 17.0 |
| Grand Total | 188 |
CY 2007 Reports (1 Jan—3 Aug)
| Report Source | Number | Percent |
|---|---|---|
| Mfr. | 46 | 67.6 |
| Direct | 10 | 14.7 |
| MedSun | 8 | 11.8 |
| Others | 4 | 5.9 |
| Blank | 0 | 0 |
| Grand Total | 68 |
Reported Adverse Reactions (1 January—3 August 2007)
|
|
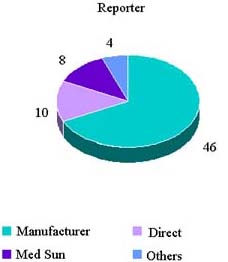
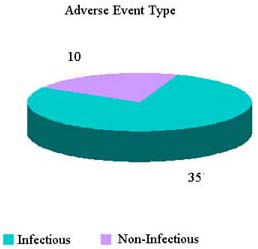
HCT/P Deviation 21 CFR 1271.3(dd)
- An event that represents a deviation from applicable regulations, standards or established specifications that relate to prevention of communicable disease transmission or contamination; or
- An unexpected or unforeseeable event that may be related to transmission or potential transmission of a communicable disease or may lead to HCT/P contamination
HCT/P Deviation Reporting 21 CFR 1271.350(b)
- Required for “361”nonreproductiveHCT/Ps recovered on or after 5/25/05
- Deviations related to a distributed HCT/Ps
- All deviations must be investigated
- Only report deviations related to “core” CGTPs (1271.150(b))
- Must report those that occurred in your facility or in a facility that performed a manufacturing step for you under contract, agreement or other arrangement
When, How, Where
- As soon as possible, not to exceed 45 days after discovery
- www.fda.gov/cber/biodev/biodev.htm
- Form FDA-3486 and instructions
- Electronically or by mail
- Indicate HCT/P Deviation Codes
HCT/P Deviation Codes
| DE | Donor Eligibility |
|---|---|
| DS | Donor Screening |
| DT | Donor Testing |
| EC | Environmental control |
| SR | Supplies and reagents |
| RE | Recovery |
| PC | Processing |
| LC | Labeling control |
| ST | Storage |
| SD | Receipt, Pre-distribution Shipment, Distribution |
| Reportable HCT/P Deviations | FY ‘05 | FY ‘06 | FY ‘07 |
|---|---|---|---|
| Donor Eligibility | 8 | 32 | 20 |
| Donor Screening | 12 | 5 | |
| Donor Testing | 33 | 52 | |
| Environmental Control | 1 | 2 | |
| Supplies and Reagents | 3 | 6 | |
| Recovery | 2 | 5 | |
| Processing | 14 | 15 | |
| Labeling Control | 1 | 2 | 1 |
| Storage | 1 | ||
| Receipt, Pre-Dist., Dist. | 4 | 43 | 28 |
| Not reportable | 15 | 77 | 40 |
| Total | 28 | 220 | 176 |
FY ’07 10-1-06 to 7-31-07
HCT/P Deviation Reports (Reportable and Not Reportable)
FY ’2007 (FY07=10/1/06-7/31/2007)
- Musculo-skeletal: 1
- Skin: 7
- Ocular: 35
- Hematopoietic stem cell: 120
- Somatic cells/leukocytes: 1
- Reproductive: 1
Inspection Information
FY 06 HCT/P Inspections Accomplished
| Type of HCT/P establishment | # Inspections Accomplished |
| Reproductive tissues | 87 |
| Cord blood stem cells Peripheral blood stem cells | 36 |
| All other HCT/Ps 9e.g. musculoskeletal, ocular, recovery, distributors) | 234 |
| Total | 354* |
| Reportable HCT/P Deviations | FY ‘05 | FY ‘06 | FY ‘07 |
|---|---|---|---|
| Donor Eligibility | 8 | 32 | 20 |
| Donor Screening | 12 | 5 | |
| Donor Testing | 33 | 52 | |
| Environmental Control | 1 | 2 | |
| Supplies and Reagents | 3 | 6 | |
| Recovery | 2 | 5 | |
| Processing | 14 | 15 | |
| Labeling Control | 1 | 2 | 1 |
| Storage | 1 | ||
| Receipt, Pre-Dist., Dist. | 4 | 43 | 28 |
| Not reportable | 15 | 77 | 40 |
| Total | 28 | 220 | 176 |
FY 06 HCT/P Inspections Accomplished
| Type of HCT/P establishment | # Inspections Accomplished |
|---|---|
| Reproductive tissues | 87 |
| Cord blood stem cells Peripheral blood stem cells | 36 |
| All other HCT/Ps (e.g. musculoskeletal, ocular, recovery, distributors) | 234 |
| Total | 354+ |
* Sum of individual inspections do not equal total (354) due to two inspections that were conducted for products in multiple categories
FY 07 HCT/P Inspections Accomplished*
| Type of HCT/P establishment | # Inspections Accomplished |
|---|---|
| Reproductive tissues | 101 |
| Cord blood stem cells Peripheral blood stem cells |
15 |
| All other HCT/Ps (e.g. musculoskeletal, ocular, recovery, distributors) |
207 |
| Total | 323 |
*as of July 5, 2007
FY 06 HCT/P Inspection Classifications
| Type of HCT/P establishment | NAI | VAI | OAI |
|---|---|---|---|
| Reproductive tissues | 55 | 26 | 5 |
| Cord blood stem cells Peripheral blood stem cells | 24 | 11 | 0 |
| All other HCT/Ps (e.g. musculoskeletal, ocular, recovery, distributors) | 170 | 59 | 5 |
| Total | 249 | 96 | 10 |
FY 07 HCT/P Inspection Classifications*
| Type of HCT/P establishment | NAI | VAI | OAI |
|---|---|---|---|
| Reproductive tissues | 66 | 29 | 6 |
| Cord blood stem cells Peripheral blood stem cells |
13 | 2 | 0 |
| All other HCT/Ps (e.g. musculoskeletal, ocular, recovery, distributors) |
150 | 54 | 3 |
| Total | 229 | 85 | 9 |
*as of July 5, 2007
FY06 HCT/P Inspection Results
- Less than 30% of HCT/P inspections resulted in issuance of Form FDA-483s
- Regulatory Actions Issued in FY06
- 2 Orders to Cease Manufacture and Retain HCT/Ps – Recovery establishments
- 1 Warning Letter – Reproductive
- 2 Untitled Letters – Reproductive
FY07 HCT/P Inspection Results*
- Less than 30% of HCT/P inspections resulted in issuance of Form FDA-483s
- Regulatory Actions Issued in FY07
- 2 Warning Letter
- 1 Reproductive establishment
- 1 Testing Laboratory
- 2 Untitled Letters
- 2 Reproductive establishments
*as of July 5, 2007
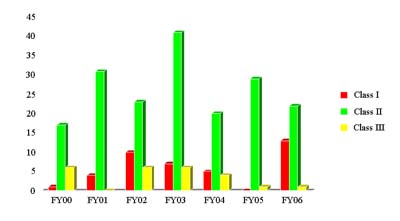
Classified Recalls FY 2006
| HCT/Ps | CBER Total (all products) |
|
| Class I | 13 | 13 |
| Class II | 22 | 1377 |
| Class III | 1 | 451 |
HCT/P Recalls Classified
CGTPs
- Draft guidance in progress
- Contains Q and As from various industry groups
- Guidance for Industry: Compliance with 21 CFR 1271.150(c)(1) Manufacturing Arrangement, September 2006
- Discusses your responsibility for ensuring the establishments with which you have manufacturing arrangements are in compliance with CGTP requirements
- Provides examples for doing so
http://www.fda.gov/cber/gdlns/cgtpmanuf.htm
Draft Cord Blood Guidance
- Published 1/17/07, comments were due by 4/17/07
- Recommends a path to licensure for establishments that manufacture cord blood for specified indications (hematopoietic reconstitution in patients with hematologic malignancies)
Draft Cord Blood Guidance
- Describes FDA's approach to the regulation of cord blood hematopoieticstem/progenitor cells that are:
- Minimally manipulated
- Used to replenish the bone marrow in patients with blood-related malignancies and
- Used in recipients unrelated to the donor of the stem cells
Draft Cord Blood Guidance
- Rather than submitting their own clinical data, manufacturers may cite existing data in the docket
- Guidance also provides recommendations for
- Content and format of information to submit with an application
- Manufacturing cord blood products
- Compliance with applicable regulatory requirements
Draft Cord Blood Guidance
- Was discussed at Cellular, Tissue and Gene Therapies Advisory Committee on March 30, 2007
- http://www.fda.gov/cber/advisory/ctgt/ctgt0307.htm
Guidance: HCT/Ps Tested Using Pooled Specimens or Diagnostic Tests
- Published January 24, 2007
- For immediate implementation
- Comments to the docket accepted
- Applies to HCT/Ps recovered after 5/25/05 to 30 days after publication http://www.fda.gov/cber/gdlns/hctppool.htm
Guidance: HCT/Ps Tested Using Pooled Specimens or Diagnostic Tests
- Addresses:
- Distributed HCT/Ps and those in inventory
- Need for HCT/P deviation reports if distributed
- Retesting/labeling for future distribution of HCT/Ps in-inventory
Testing Guidance – cont.
- FDA is exercising enforcement discretion to allow distribution of these HCT/Ps
- Limited to the two testing deficiencies
- Pertains only to recently regulated living donors (hematopoietic stem/progenitor cells and reproductive cells and tissues)
- For distributed HCT/Ps, deviation reports only required for hematopoietic stem cells from first or second degree blood relatives
- One report for all affected HCT/Ps
Testing Guidance: In Inventory HCT/Ps—Retesting Before Distribution
- Recommended using original donor specimen
- As long as it can be tested in accordance with the manufacturer’s instructions
- New specimen OK
- Distribute “as is” if negative/nonreactive
Testing Guidance: In Inventory HCT/Ps—Retesting Before Distribution
- If retesting not feasible
- Document reason that retesting is not feasible in accompanying records and in files
- Physician notification
- Labeled “WARNING: Advise patient of communicable disease risks”
Testing Guidance: Retesting Donors
- You can distribute in-inventory HCT/Ps from the following donors, even if you are unable to retest them
- Hematopoietic stem cells (non-autologous)
- Cryopreserved embryos formed using 3rdparty gametes
- If you cannot retest anonymous or directed donors of cryopreservedsemen or oocytes, you cannot distribute
Blood Vessel Final Rule
- Final Rule published in March 2007 and is effective 30 days after publication
- Revises 21 CFR Part 1271.3(d) for articles not considered HCT/Ps
- New 1271.3(d)(8): Blood vessels recovered with an organ, as defined in 42 CFR 121.2, that are intended for use in organ transplantation and labeled “For use in organ transplantation only”
- Revises 42 CFR to make these blood vessels “part of”organs
Blood Vessel Final Rule
- This was a joint rule by FDA and HRSA
- Addresses blood vessels recovered with organs and intended for use in organ transplantation
- Health Resources and Services Administration (HRSA) oversight
Donor Eligibility Guidance
| Guidance for Industry Eligibility Determination for Donors of Human Cells, Tissues, and Cellular and Tissue-Based Products (HCT/Ps) |
- Reposted as Level II Guidance (minor changes) 8 August 2007 Changes include:
- Typographical changes
Donor Eligibility Guidance
- III. J. ---The description of accompanying records for quarantine shipping under §1271.60(c) now reads: Identify the donor (e.g., by a distinct identification code affixed to the HCT/P container); removed “but not by name, social security number, or medical record number (except in the case of an autologous, or directed reproductive donors, or donations made by first-degree or second-degree blood relatives §1271.55(a)(1))”
Donor Eligibility Guidance
- IV. E. 3. ---Persons with hemophilia or other related clotting disorders who have received human-derived clotting factor concentrates in the preceding 5 years (risk factor for HIV, Hepatitis B and Hepatitis C). A person who has received human-derived clotting factor concentrates only once may be eligible to donate.
Donor Eligibility Guidance
- IV. E. 18. --- The following example was added to the chlamydia and gonorrhea donor screening criteria:
A potential donor has a medical record indicating that she was treated record indicating that she was treated for Chlamydia 14 months ago. No for Chlamydia 14 months ago. No followfollow--up testing was performed at the up testing was performed at the time of treatment. The medical record time of treatment. The medical record serves as evidence that she received serves as evidence that she received treatment more than 12 months ago.treatment more than 12 months ago.
Donor Eligibility Guidance
Continued--Since the medical record does not include information that a follow-up test was performed after treatment, and was negative, there is no evidence that the treatment was successful. However, a current negative test for Chlamydia (as part of the current donor testing) may serve as evidence that the treatment that occurred more than 12 months ago was successful.
Donor Eligibility Guidance
- IV. E. Note under 28.
- Clarification of HIV Group O screening–Establishments utilizing an HIV-1/2 antibody donor screening test that has been licensed by FDA and is specifically labeled in the Intended Use Section of the package insert as sensitive for detection of HIV group O antibodies may delete items 27 and 28 from their screening procedures.
Items 27 and 28 refer to screening questions pertaining only to HIV group O.
Donor Eligibility Guidance
- If such establishments continue to ask items 27 and 28, the donor eligibility may be based on the results of the donor screening test regardless of the answers to items 27 and 28. Establishments that do not utilize an HIV antibody donor screening test that has been licensed by FDA for detection of HIV group O antibodies should continue to ask these items.
www.fda.gov/cber/tissue/prod.htm
www.fda.gov/cber/products/testkits.htm
Items 27 and 28 refer to screening questions pertaining only to HIV group O.
Donor Eligibility Guidance
- In the clinical evidence and physical evidence sections, there are changes to make the use of the word “unexplained” before both “hepatomegaly”and “oral thrush”to be consistent
- IV. F. 2. ---Added “unexplained”before “hepatomegaly”
- IV. G. 7. ---Added “unexplained”before “oral thrush”
Donor Eligibility Guidance
- IV. F. 7. ---Clinical evidence for HTLV clarified: HTLV infection (Screening and donor deferral for HTLV required only for viable, leukocyte-rich HCT/P donors)
Donor Eligibility Guidance
- V. D. ---The specimen for testing from the birth mother must be collected within seven days of donation by the infant (§1271.80(b)), unless the donation consists of peripheral blood stem/progenitor cells or bone marrow according to 1271.80(b)(1).
Donor Eligibility Guidance
- VI. A. 5. ---Information about syphilis testing updated—Treponema pallidum (FDA-cleared screening test for syphilis or FDA-cleared diagnostic serologic test for syphilis6)
- 6For purposes of this guidance, we consider FDA-cleared diagnostic serological tests to be adequate for use in donor screening for syphilis.
Human Tissue Task Force
- Formed in August to evaluate Formed in August to evaluate effectiveness of new regulations and to effectiveness of new regulations and to identify whether additional steps identify whether additional steps necessary to strengthen regulatory necessary to strengthen regulatory framework
- Collaborative effort between CBER, Collaborative effort between CBER, ORA and OCORA and OC
Human Tissue Task Force
- Memo containing recommendations provided to CBER and ORA senior management
- Report posted June 12, 2007
- More details re: HTTF from Dr. St. Martin

Contact Information
Melissa A. Greenwald, M.D.
1401 Rockville Pike HFM-775
Rockville, MD 20852
- Email CBER:
- Manufacturers:
matt@cber.fda.gov - Consumers, health care
octma@cber.fda.gov - Phone:
301-827-2000