Office
of Drug Safety
Annual Report
Center for Drug Evaluation
and Research
Food and Drug Administration
Annual Report
FY 2001
EXECUTIVE
SUMMARY
INTRODUCTION
I. OFFICE VISION
BUILDING
II. CURRENT OPDRA STRUCTURE AND
FUNCTION
III. OFFICE OF DRUG SAFETY: RATIONALE
FOR REORGANIZATION
IV. SAFETY REVIEW OF DRUGS
Overview
Drug Risk Assessment: From Signals to
Regulatory Actions
Pre-approval Safety Conferences
Waivers of Postmarketing Safety
Reporting Requirements
V. RISK ASSESSMENT
AERS Reports
MedWatch's Central Triage Unit
(CTU)
Electronic Submission of Adverse
Experiences Initiative
AERS
DataMart
VI. RISK MANAGEMENT/COMMUNICATION
DDRE Accomplishments
Plans for the Future
VII. MEDICATION ERRORS
Pre-approval Assessments
Post-approval evaluation/signal
detection
Plans for the Future
VIII. EXTRAMURAL RESOURCE DEVELOPMENT
Cooperative Agreement Program
New Data Initiatives
Drug Use Data Resources
IX. INTERNATIONAL ACTIVITIES
International Conference on
Harmonization (ICH)
Video-conferences
International Visitors
AERS to Canada
X. PATIENT SAFETY INITIATIVES
QuIC responses to IOM report
Patient Safety Task Force
Other Safety Initiatives
XI. RISK MANAGEMENT
Introduction
Risk Management Toolbox
Advisory Committee Support
Risk Management White Papers
Drug Safety Seminar Series
Fellowship Program
XII. LEVERAGING AND OUTREACH
Accomplishments
XIII. REGULATORY RESEARCH
XIV. REGULATIONS, GUIDANCES AND MAPPS
CONCLUSION
APPENDIX 1 - OPDRA ORGANIZATIONAL
CHART
APPENDIX 2 - OPDRA MEETING
PRESENTATIONS
APPENDIX 3 - OPDRA PUBLICATIONS
APPENDIX 4 - POSTERS
 Back to Top
Back to Top
ANNUAL REPORT - 2001
Executive
Summary:
Fiscal Year 2001 was a successful
year for OPDRA in terms of achieving organizational, strategic, regulatory and
scientific objectives. These accomplishments, which are described in detail
later in this report, are summarized below:
· Planned the reorganization of the
Office of Drug Safety to incorporate MedWatch and Patient Labeling program
functions and to achieve additional strategic objectives.
· Supported Federal patient safety
and medication error initiatives.
· Engaged in extensive leveraging
activities with other Federal agencies, private organizations, industry and
the Centers for Education and Research on Therapeutics (CERTs) to support our
mission of ensuring the safety of drugs.
· Continued the development and
enhancement of the Adverse Event Reporting System (AERS).
· Completed Step 4 signoff of E2B
and continued progress in developing other aspects of the International
Conference on Harmonization (ICH) that relate to postmarketing adverse event
reporting and drug safety.
· Increased support of the Office of
Review Management regulatory actions and risk management program development.
· Acquired mission-critical data
resources including longitudinal, inpatient and pediatric inpatient data
utilization data.
· Maximized the utilization of
managed care databases through the cooperative agreements to evaluate
important safety questions in formal epidemiological studies.
· Recompeted the cooperative
agreement grant through the competitive RFA process to ensure continued access
to these important databases by FDA for the coming years.
· Completed and published important
research in quantifying the effect of regulatory actions.
· Codified and expanded
collaboration with other regulatory drug bodies.
As the new year begins, there are
several new challenges to meet and strategic initiatives to plan.
The first priority for the new Office
will be the tactical implementation of the reorganization of the Office of
Drug Safety (ODS); including selecting appropriate people to lead the
different ODS divisions and establishing internal procedures for existing and
newly assigned responsibilities (e.g. MedWatch and patient labeling). We will
also have to develop processes to incorporate new data resources into the
business of safety evaluation.
A second, but equally important
priority, is developing ODS/Office of Review Management (soon to be the Office
of New Drug Evaluation) interaction operating principles. It is critical that
ODS maintains close communication and synergistic relationships with ORM and
the ORM reviewing divisions. Codification of relationships through MaPPs on
such issues as tradename reviews, postmarketing safety issues, evaluation of
risk management programs and medication guides will be addressed. Furthermore,
the shared responsibilities in the review of Periodic Safety Update Reports (PSURs)
will be addressed through a MaPP and the initiation of development of a
guidance to industry. The appropriate use of the new Drug Safety and Risk
Management Advisory Committee will also be codified in a MaPP. In addition to
these MaPPs and guidances, it is critically important for ODS to maintain
regular participation in ORM activities by attending critical ORM meetings
such as ministaff, division director meetings, etc.
The final priority for the new office
will focus on the Center initiative relating to Risk Management. The
development of the Risk Management White Paper, the identification of a risk
management and risk communication research agenda, the successful launch and
utilization of the new Drug Safety and Risk Management Advisory Committee, the
optimal use of MedWatch and the CERTs for risk communication and outreach, the
identification of additional data resources to optimize risk assessment, and,
ultimately, the codification of ODS' role in risk management for marketed
drugs will represent major opportunities and challenges for the next year.
Back to Top
Introduction
FY 2001 has been a challenging and
rewarding time for the Office of Postmarketing Drug Risk Assessment. In
addition to the traditional role of postmarketing assessment of adverse event
reports for marketed drugs, the Office has engaged in many initiatives focused
on developing a comprehensive risk assessment and management program that can
support ongoing Center priorities in these areas. We are very proud that CDER
management has recognized the contributions that OPDRA has made and is
recommending increasing OPDRA's responsibilities in a proposed reorganization.
This decision is based on a growing sense of confidence that OPDRA has worked
effectively in the past to ensure the safer use of drugs, thereby; better
protecting the public health. The purpose of this annual report is to
highlight the OPDRA accomplishments over the past year and to outline
strategic plans for the upcoming year.
I. Office Vision Building:
The success over the recent past has
significantly improved morale and sense of purpose in OPDRA. We have worked
with an external consultant to identify critical areas to be addressed by
management and staff to further improve communication and working
relationships within the office. We believe the following vision is a good
foundation on which to continue to build the best possible postmarketing drug
safety program for CDER and FDA.
Vision
"OPDRA excels in the
assessment, management and communication of drug risks. OPDRA integrates
science, research, and the principles of public health to guide appropriate
and consistent regulatory decision-making in order to optimize the safe use of
marketed drugs. OPDRA is a growing, evolving, and adaptable unit based on
outstanding individual and team efforts and we recognize the value and
contributions of all members of our organization."
Back to Top
II. Current OPDRA Structure and
Function:
OPDRA, as originally established in
1998, consists of an Immediate Office with two Divisions of Drug Risk
Evaluation (DDREs) and various staff units to support medication error
initiatives and resource management. Peter Honig, M.D., and Martin Himmel,
M.D. are the OPDRA Director and Deputy Director, respectively.
The Divisions of Drug Risk Evaluation
led by Drs. Julie Beitz (Director) and Anne Trontell (Deputy Director)
collaborate with CDER's new drug review offices to ensure that a careful
safety evaluation of marketed drugs is provided. In this role, OPDRA's safety
evaluators and epidemiologists critically review adverse experience reports
that are submitted by manufacturers, health care professionals and consumers.
The DDREs are proactively engaged in important FDA functions and participate
in advisory committee preparation, risk management program development and
pre-approval safety conferences. OPDRA increasingly interacts with our
regulatory counterparts in Australia, Canada and the European Union to share
information about Adverse Drug Reaction (ADR) reports and drug safety issues.
OPDRA's Medication Error Staff of
eight safety evaluators assesses risk factors that could lead to medication
errors resulting from nomenclature, labeling and packaging of drug products.
This staff, led by Captain Jerry Phillips reviews proposed proprietary names
for unapproved drug products and is responsible for assessing not only actual
medication errors that have led to patient injury or death, but also those
errors that have the potential to cause patient harm.
OPDRA's Information Technology Staff
led by Jerry Phillips assume a wide array of responsibilities including
desktop support, AERS special searches, database development and support, and
website maintenance.
The Extramural Program Staff, led by
Jerry Phillips, is critical in maintaining existing grants and contracts as
well as identifying and procuring new grants and data resources.
Back to Top
III. Office of Drug Safety: Rationale
for Reorganization
OPDRA has worked hard to build strong
relationships with CDER staff and other OPDRA customers by creating
trust and demonstrating value. CDER management has proposed a significant
reorganization of the CDER structure that includes increasing OPDRA's
responsibilities and retitling it the Office of Drug Safety. This is a
tremendous honor that has been accompanied with an increase in resources and
reflects the value that OPDRA has contributed to CDER. ODS will incorporate
the MedWatch and patient labeling responsibilities that are currently in other
parts of the Center. In order to meet this challenge, the Office of Drug
Safety will be reorganized to meet these challenges. The guiding principles
behind the reorganization include:
· Provide staff development
opportunities
· Harness economies of scale and
configure to maximize efficiency
· Allow synergy of the new
responsibilities and existing functions and provide avenues of communication
for these interactions
· Create a scalable structure that
could allow assumption of new responsibilities in the future
· Allow for localized
decision-making
· Strongly support the scientific
base for risk assessment, management and communication
The reorganization will merge the
existing DDREs into one division that takes advantage of economies of scale
and efficiencies. Dr. Julie Beitz will direct this large division which will
remain a centerpiece of the Office of Drug Safety. The second division will be
titled the Division of Medication Errors and Technical Support (DMETS) and be
headed by Jerry Phillips. Finally, a third division will be created and led by
Dr. Anne Trontell. This new division will be called the Division of
Surveillance, Research and Communication Support (SuRCoS) and assume the
leadership of new responsibilities (MedWatch, MedGuides/patient labeling) and
identification and management of an increasing array of postmarketing data
resources.
Back to Top
IV. Safety Review of Drugs
Overview
DDRE twenty safety evaluators
performed hands-on review of adverse drug event reports reviewed by FDA
throughout the year. This was carried out through daily review of AERS reports
that are triaged to each Safety Evaluator's electronic inbox. These reports
include 15-day expedited reports, direct serious reports, and reports of
designated medical events. The latter are serious events targeted by OPDRA
that are often drug-related (e.g., liver failure, bone marrow failure,
Stevens-Johnson syndrome, etc). Individual reports may trigger further
evaluation of similar reports in the database and development of a case
series. Case series could signal important safety concerns prompting
regulatory actions both in the U.S. and abroad.
A total of 634 consults were received
and processed in fiscal year 2001 by the Divisions of Drug Risk Evaluation I
and II. The origin of these consult requests was as follows:
355 consults (56%) originated from
the fifteen ORM Review Divisions;
145 consults (23%) were generated
by OPDRA staff;
134 consults (21%) originated from
requests from other sources.
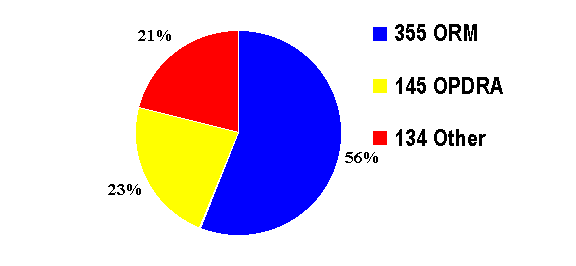
Summary data are presented
graphically by requestor (ORM review division, self-generated, other sources)
and by review division.
Consults
by HFD Division
Back to Top
Other consult requests from within
FDA include: Office of the Commissioner, CFSAN, CDRH, CBER, Office of the
Center Director, Office of Criminal Investigations, Office of Compliance,
Office of General Counsel, Office of Women's Health, Office of Legislative
Affairs, Regulatory Policy Staff, Office of Generic Drugs, Office of
Pharmaceutical Sciences, Office of Clinical Pharmacology and Biopharmaceutics,
Controlled Substances Staff, MedWatch, Pregnancy Working Group, Executive
Projects Team, and Office of Training and Communications.
Consult requests from outside of FDA
include: Congress, GAO, the Secretary of DHHS, World Health Organization,
Federal Bureau of Investigation, Department of Justice, Centers for Disease
Control, U.S. Consumer Products Safety Commission, University of Pennsylvania
(CERTS Program), VA Medical Center (California), Division of Federal State
Relations, and several Foreign Regulatory Agencies.
Drug Risk Assessment: From Signals to
Regulatory Actions
OPDRA staff worked closely with
review division staff to develop the scientific basis for labeled warnings,
postmarketing safety studies, drug withdrawals and compliance activities. The
following are selected regulatory actions that were supported in OPDRA:
Boxed Warnings
· Gemtuzumab ozogomicin: new boxed
warnings for serious hypersensitivity reactions, adult respiratory distress
syndrome, veno-occlusive disease.
· Capecitabine/warfarin interaction:
new boxed warning added
· Itraconazole - boxed warning
describing the risk of congestive heart failure with itraconazole when used
for the treatment of onychomycosis; a Public Health Advisory issued May 2001.
Other Important Labeled Warnings
· Topiramate: warning for glaucoma
· Terbinafine: warning for liver
failure; a Public Health Advisory issued May 2001
· Linezolid: OPDRA identified
myelosuppression as a safety signal; this resulted in a new labeled warning
and a Dear Health Care Professional Letter was issued by the sponsor
Postmarketing Safety Studies
· Perflutren: a postmarketing
surveillance study to monitor adverse events in at least 1000 patients exposed
to this ultrasound drug product was developed
· Mifepristone: OPDRA was
instrumental in crafting Phase 4 commitments in conjunction with the drug's
marketing approval, and reviewing the submitted post approval protocols and
foreign postmarketing report summaries.
Back to Top
Drug Withdrawals
· Cerivastatin: OPDRA coordinated
with the review division and shared scientific developments with our foreign
regulatory counterparts regarding rhabdomyolysis associated with cerivastatin.
This adverse event was the primary reason for which Bayer withdrew
cerivastatin from the global market.
· Alosetron: OPDRA's review of
postmarketing adverse event reports supported the review division's concerns
regarding ischemic colitis and serious constipation. The sponsor voluntarily
stopped marketing Alosetron in November 2000.
Pre-approval
Safety Conferences
Pre-approval safety conferences are
held with review division staff as per CDER MaPP 6010.1 prior to the marketing
approval of a new drug product. These meetings typically involve discussions
of safety information in labeling. Specific safety concerns are identified
that OPDRA staff will monitor after product launch. Other topics may include
the need for postmarketing safety studies, patient registries, or a risk
management program to closely monitor safety concerns. Examples of recently
approved drugs and safety issues discussed at recent pre-approval safety
conferences are:
· Ziprasidone: need for OPDRA to
continuously monitor spontaneous reports of QTc prolongation and related
events, with quarterly updates to the review division
· Nasiritide: need for a warning
for fluid retention in labeling
· Bosentan: need for boxed
warnings in labeling for teratogenicity and liver toxicity; need for a
medication guide and risk management programs to monitor pregnancy exposures
and cases of liver injury after product launch
· Bimatroprost and Travoprost ophthalmic
solutions: approved as second line treatments for glaucoma
(instead of first line) due to concerns about cardiovascular events and
deaths with a related product (latanoprost)
· Verteporfin:
need for information on drug-drug interactions in labeling
Back to Top
Waivers of
Postmarketing Safety Reporting Requirements
OPDRA has the authority to grant
waivers of postmarketing safety reporting requirements (additional information
may be found at
http://www.fda.gov/cder/mapp/6004-1.pdf).
In addition, OPDRA is developing a database for waiver information that will
accessible to CDER.
In FY 2001, OPDRA staff extensively
briefed CDER staff about the waiver procedures.
OPDRA encourages industry to request
a waiver of the requirement to submit FDA form 3500A for each adverse
experience that is determined to be both nonserious and labeled. By the end of
FY 2001, over 60 firms had been granted this type of waiver for over 2300
applications (NDAs and ANDAs).
OPDRA has granted waivers to firms of
requirements to submit postmarketing periodic safety reports in the format
described in the regulations (21 CFR 314.80). Instead, firms may use the
Periodic Safety Update Report (PSUR) format described in ICH E2C (with
modifications to meet U.S. requirements).
When a firm asks to modify the
frequency of submission of a report, OPDRA consults with the appropriate
review division before acting on the waiver request. By the end of FY 2001,
fewer than 10 firms had been granted PSUR waivers (for PSUR format with or
without frequency changes) for fewer than 25 applications. However, through
OPDRA presentations to industry, firms are becoming aware of this option, and
the number of requests for this type of waiver is rising dramatically. By
mid-November 2001, OPDRA had received requests from more than 15 firms for
more than 275 applications.
Contact Person: Kathleen Bongiovanni,
301-827-3191
Back to Top
V. Risk Assessment
AERS Reports
OPDRA's AERS group oversees
activities relating to the adverse event reporting system, including paper and
electronic submission of adverse event reports. The group provides the
programmatic expertise to the AERS database application, serving to bridge the
users with the Office of Information Technology for daily operations, system
troubleshooting and long-range improvements. The group also serves as the
primary liaison between CDER and foreign regulatory bodies regarding drug
safety issues. In addition, the group provides expertise on the Medical
Dictionary for Regulatory Activities (MedDRA), an ICH-developed medical
terminology used for coding medical information and which is used in AERS.
In FY2001, AERS received 205,101
Individual Safety Reports (ISRs) + 75,989 non-serious periodic ISRs (Total =
281,090)1 . At the present time, AERS is
entering reports for direct2, 15-day
(expedited)3 and serious periodic4
ISRs. AERS captures all of the ISRs for current New Molecular Entities (NMEs).
The chart below shows the reporting trend over time. It is anticipated that
the total number of reports in CY 2001 will be close to 271,000 ISRs.
CDER Post-Marketing Adverse
Events Reports Received (Data in Excel
Spreadsheet)
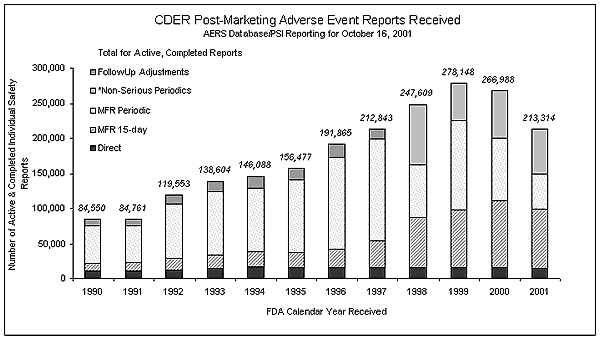
If this trend holds, it will mark the
second consecutive year in which the number of reports entered into AERS has
slightly decreased. There are several possible explanations for this including
the decrease in the number of NMEs approved by the center since the high water
mark in 1996 as well as the aggressive OPDRA waiver program relieving the
industry from the requirement of submitting non-serious and labeled events.
Back to Top
The table below indicates the top 20
suspect products submitted to FDA and entered in AERS (NME is defined as a
drug approved in the past three years and designated by an asterisk *)
Rofecoxib (Vioxx) 6,994*
Etanercept (Enbrel) 6,789*
Isotretinoin (Accutane) 3,605
Alosetron HCl (Lotronex) 3,190*
Celecoxib (Celebrex) 3,087*
Sertraline HCl (Zoloft) 2,834
Atorvastatin Ca (Lipitor) 2,641
Omeprazole (Prilosec) 2,066
Cerivastatin Na (Baycol) 1,992
Rosiglitazone (Avandia) 1,965*
Citalopram HBr (Celexa) 1,947*
Infliximab (Remicade) 1,635*
Sildenafil (Viagra) 1,642*
Orlistat (Xenical) 1,633*
Tolterdine (Detrol) 1,515*
Olanzapine (Zyprexa) 1,480
Pioglitazone HCl (Actos) 1,464*
Azithromycin (Zithromax)1,413
Thalidomide (Thalmid) 1,378*
Back to Top
MedWatch's Central Triage Unit
(CTU):
OPDRA staffs the MedWatch Central
Triage Unit that supports the identification and appropriate routing of direct
reports on all medical products received through MedWatch. Two FTEs are
allocated to this important function. In FY01, CTU processed 23,601 MedWatch
reports for all of FDA's centers. As can be seen below, 70% of the reports
were triaged to CDER and entered into AERS. Direct reports from biologic
therapeutics are also entered into AERS.
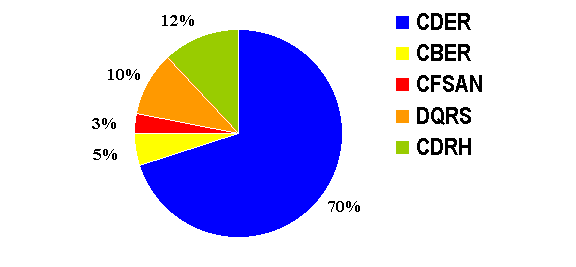
The Central Triage Unit also tracks
the reports by the source of the submission and seriousness of the adverse
experience. As can be seen from the figure below, the single-leading source of
direct reports to FDA are pharmacists. 59% of the reports are serious by
outcome and 30% of the reports are being submitted to the MedWatch are via the
internet
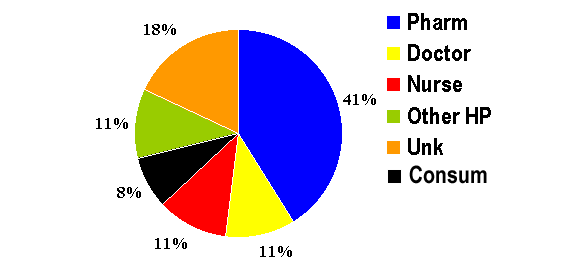
Back to Top
Electronic Submission of Adverse Experiences Initiative:
This is a high priority for OPDRA and
CDER because the successful implementation of electronic submissions will
result in considerable cost savings to CDER and FDA. To date, the electronic
submissions (e-sub) pilot has been very successful in establishing a mechanism
for companies to work with FDA in sending e-sub reports into AERS.
CDER implemented an Electronic
Submission Product Test Pilot for AERS in October 2000. This pilot provided a
mechanism for companies to test and send electronic submissions of expedited
reports via physical media or gateway (without attachments) directly into AERS
(Docket 925-0251).
In spring of 2002, AERS will upgrade
the Data Transfer Document (DTD) to 2.1 and always maintain the latest two
versions of DTD. 11,404 individual case safety reports (ICSR)5
were submitted electronically under the pilot program in FY 2001.
The current industry participants in
e-sub include: Bristol-Myers Squibb, GlaxoSmithKline, Merck, Roche, Pharmacia,
Sanofi-Synthelabo, Pfizer, MedImmune, Dupont Pharmaceuticals, J&J Pharma
Group, Eli Lilly, AstraZeneca, Immunex, Celgene, Bayer Vital Pharmaceuticals
(Germany).
AERS can accept reports via the
gateway or physical media without attachments (Docket 925-0251). In January
2002, AERS will upgrade the Data Transfer Document (DTD) to 2.1 and always
maintain the latest two versions of DTD.
AERS information is made publicly
available by providing quarterly CD ROMs in ASCll format and SGML (ICH E2B)
format for six AERS data tables. The tables include demographics, drugs,
reactions, report source, outcomes, and therapy date data. The CD ROMs are
provided to the Office of the Commissioner for publications. The National
Technical Information Service (NTIS) copies the CDs and offers them for sale
to the public. The Data are currently not made publicly available through the
FOI or Web because of resource limitations.
AERS
DataMart:
One of the major success stories has
been the development of the AERS DataMart tool by OIT in conjunction with
OPDRA. This has allowed CDER to eliminate the dual flow of expedited (i.e.
15-day) paper reports to OPDRA and the medical officers in the reviewing
divisions, a strategy consistent with OPDRA's responsibility for the review
and aggregate analysis of these reports while at the same time allowing the
reviewing medical officer in ORM to have access to the information contained
in these reports and in AERS. Medical officers are notified via email that an
expedited report has been received and can be viewed in the DataMart desktop
application that also allows for aggregate analyses of such events.
Contact person: Roger Goetsch, Pharm.
D., 301-770-9299
Back to Top
VI. Risk Management/Communication
DDRE Accomplishments
OPDRA staff worked closely with
review division staff to develop and negotiate the following comprehensive
programs designed to manage known risks of drug products.
· Isotretinoin (Accutane): a
pregnancy prevention program including development of metrics to assess the
program's effectiveness was approved October 31, 2001; development of a
Medication Guide (issued January 2001) and revised informed consent to alert
patients of the risks of depression and suicide; need for OPDRA to
continuously monitor spontaneous reports of suicide and depression
· GHB (Xyrem): a restricted
distribution program involving patient and prescriber registries and a central
pharmacy to limit illicit use after product launch was discussed at an FDA
Advisory Committee
· Bosentan (Tracleer): a restricted
distribution program to ensure monitoring for liver toxicity and pregnancy
prevention was established at the time of marketing approval on November 20,
2001
Advisory Committee Preparation: OPDRA
provides pivotal support to review divisions at FDA Advisory Committee
Meetings. OPDRA staff has provided testimony on drug-related safety issues,
prepared briefing materials for Committee Members and participated in
Committee discussions. Examples of recent Advisory Committee Meetings include:
· Phenylpropanolamine (PPA) and risk
of hemorrhagic stroke: OPDRA staff performed a critical review of the
Hemorrhagic Stroke Project study and testified before the Non Prescription
Drugs Advisory Committee. The Committee recommended that PPA not be marketed
OTC. FDA issued a Public Health Advisory which led to a voluntary withdrawal
of PPA from the US and Canadian markets.
· Cox-2 inhibitors: cardiovascular
and thrombotic events, gastrointestinal hemorrhagic deaths, and renal adverse
events were evaluated by OPDRA staff; written briefing materials were
prepared. The Advisory Committee recommended labeling cardiovascular risks for
rofecoxib.
· OTC Switch of Non-sedating
Antihistamines: OPDRA and review division staff prepared a comprehensive
review of several safety concerns including ventricular arrhythmias, seizures,
and drug-drug interactions for loratadine, fexofenadine, cetirizine. The
Committee recommended that all three products were appropriate for OTC use.
· Telithromycin: OPDRA staff
testified on the risks of QTc prolongation and torsade de pointes.
· Caspofungin: OPDRA staff testified
on the use of historical controls as a comparator group in clinical trials
submitted in support of marketing approval. The Committee recommended that use
of historical controls for this purpose was inappropriate.
Back to Top
Plans
for the Future
FDA is proposing to amend its
periodic safety reporting regulations by adding an important new type of
postmarketing periodic safety report (PSUR). The proposed content and format
for the PSUR are consistent with the ICH E2C guidance (62 FR 27470) and would
enable applicants to submit a single core document to regulatory agencies
worldwide. The purpose of a PSUR is to provide companies with a format that
has been standardized worldwide for reporting postmarketing safety information
that can be used to evaluate whether the benefit/risk ratio of a drug or
biologic has changed since market authorization.
VII. Medication Errors
During FY 2001, the Medication Error
Staff completed 305 consults for CDER. This included recommendations on 281
premarketing proprietary names reviews and 24 postmarketing medication error
consults. The program also acquired access to a proprietary name database from
Thomson and Thomson, which allows the staff to search for sound-alike and
look-alike names to the proposed proprietary names.
Pre-approval Assessments
All proposed proprietary names,
labels and labeling in CDER are reviewed by OPDRA in its pre-marketing efforts
to minimize medication errors due to similar proprietary names or confusion
related to labeling and packaging of drug products.
Post-approval evaluation/signal
detection
After the approval of drug products,
OPDRA monitors all postmarketing reports of medication errors that are related
to naming, labeling and/or packaging. Each report is classified for severity,
type and cause of error. If appropriate, OPDRA makes specific recommendations
to the appropriate reviewing divisions on changing the labeling, packaging or
proprietary name.
Contact Person: Sammie Beam, R.Ph.,
301-827-3242
Plans for
the Future
OPDRA hopes to move forward in
developing computer software to screen proposed proprietary names for
sound-alike and look-alike similarities with the existing 15,000 marketed
proprietary names in the U.S. This tool would enhance the scientific and
analytical ability of the Office to determine the potential risk of approving
a particular proprietary name.
OPDRA plans to modify AERS by
incorporating a Taxonomy of Medication Errors to enhance its ability to detect
medication errors. This taxonomy will allow the staff to perform trend
analysis based upon the numerous causes and types of medication errors, which
is currently not available through AERS.
Back to Top
VIII. Extramural Resource Development
Cooperative Agreement Program
OPDRA's cooperative agreement program
in pharmacoepidemiology provides FDA with access to population-based
pharmacoepidemiologic databases and with the ability to study important
postmarketing drug safety questions. Last year, five sites received grant
support through this program. The sites were Boston Collaborative Drug
Surveillance Program providing access to UK General Practice Research Database
(GPRD); Harvard-Pilgrim/Health Partners providing access to data from three
HMOs; Johns Hopkins School of Medicine providing access to its HIV/AIDS
database; UnitedHealth Group providing access to claims data from its
multi-state membership; and Vanderbilt University providing access to
Tennessee Medicaid.
Cooperative agreement grants are
awarded in three-year cycles and the program was recompeted this year. Funding
for this program was reduced by $0.4 million (from $1.3 million) to $0.9
million in FY 2002. The following applications will be funded at about $300K
per year: Harvard-Pilgrim/Health Partners, United Health Group and Tennessee
Medicaid. These databases will become available for CDER use in January, 2002.
The following studies were completed
in FY 2001:
· Examined the use of cisapride in
patients with contraindicated medical conditions or who concomitantly used
contraindicated medications known to increase the risk of QT-prolongation and
torsades de pointes. The drug continued to be misused despite a major
relabeling effort and issuance of a "Dear Healthcare Professional"
letter.
· Measured the effect of four
separate labeling changes with accompanying "Dear Healthcare
Professional" letters on performance of liver enzyme monitoring with
troglitazone. After four letters, monitoring was infrequent and inconsistently
performed.
· Measured (in general
population of troglitazone users) rate of hospitalized liver injury or acute
liver failure. ODPRA found (that of 7,600 patients contributing 4,000
person-years of exposure), 5 cases of hospitalized liver injury including 1
case of acute liver failure.
· Conducted a study to
determine the pattern and extent of systemic corticosteroid use in children. A
sizable number of children were shown to be exposed to long-term (>90 days)
oral corticosteroid therapy, placing them at potential risk for adverse side
effects.
· The Johns Hopkins HIV/AIDS
clinical database was used to study the question of whether there is an
association between avascular necrosis (AVN) and use of various anti-viral
agents. A non-statistically significant increasing trend in AVN was found,
however, no particular drug, drug class or combination of therapy could be
shown to confer increased risk possibly because the study was underpowered.
· The GPRD was used to study
the use of atypical anti-psychotics and development of new-onset diabetes. The
study found an increased risk of diabetes among treated patients but was
unable to determine whether this was due directly to the drugs taken or other
factors related to their use.
· Evaluated compliance with labeling
and "Dear Healthcare Professional" letters on the use of pemoline as
a first-line therapy for attention deficit disorder and on recommended
biweekly liver enzyme monitoring. The majority of pemoline use was as
first-line therapy, despite its labeled designation as second-line therapy
because of risks of acute liver failure. Liver enzyme monitoring was rarely
performed.
Back to Top
New
Data Initiatives
OPDRA has established a New Data
Initiatives working group with responsibility for identifying potential, new
or emerging data resources that might improve its capacity to evaluate drug
safety questions. The following initiatives are significant:
· In FY 2001, OPDRA established a
pilot with Kaiser Northern California, that included accessing a database
covering 6 million lives and provides information on some details of clinical
care provided to patients within this closed-model HMO. The data quality is
considered superior to that available through the cooperative agreements.
· OPDRA and Kaiser planned a
one-year pilot project to study COX-2 and NSAID use and risk of myocardial
infarction. It is hoped that one or two collaborative in-depth epidemiologic
studies will be completed in FY 2002.
· OPDRA has established an
interagency agreement with the Veteran's Administration to study avascular
necrosis (AVN) of the hip and drugs used to treat HIV/AIDS. The VA database
(18,000 patients) includes information about underlying health status, risk
factors and medical history. In this collaboration, the VA is providing data
management support, analysis dataset support, coordination of AVN
case-validation, and expert knowledge of the database and VA healthcare
system. OPDRA is supplying epidemiologic and clinical research support,
including providing expertise for protocol development and data analysis.
OPDRA is also responsible for developing the web-based interface critical to
cost-effective case validation.
Back to Top
Drug Use Data Resources
IMS
The FDA is a long-time user of IMS
Health products and services and the recompetition of this important contract
was a major accomplishment this past fiscal year. FDA was able to provide
continued access to this important data resource. The contract provides OPDRA
with access to the following databases: National Disease and Therapeutic IndexTM
(NDTI), National Prescription Audit PlusTM (NPA), Provider
PerspectiveTM (PP), and Retail PerspectiveTM (RP). While
OPDRA uses IMS data in a variety of ways, the most frequent uses include:
quantifying the number of prescriptions dispensed to the population and
obtaining demographic information on the population exposed to pharmaceutical
products. These data are also used in association with spontaneous case report
data to understand the context within which ADRs occur. When IMS data are used
in conjunction with supplemental data obtained from population-based claims or
record-linked databases, it is possible to estimate patient exposure time for
a particular drug product.
Contact person: Joslyn Swann, R.Ph.,
M.G.A., 301-827-3166
Premier,
AdvancePCS, Child Health
Corporation of America (CHCA)
Although IMS has been a reliable
resource, there were some limitations to the program. To help resolve these
limitations, OPDRA in collaboration with FDA's Office of Facilities,
Acquisitions and Central Services issued three requests for proposals (RFPs)
under the competitive bid process. The RFPs were designed to fill in three
major blindspots in CDER's understanding on how drugs are used once they are
cleared for marketing.
As a result of these efforts, CDER
now has access to three large, commercial databases that contain detailed
information on the actual use of marketed prescription drugs in hospitalized
and non-hospitalized adults as well as hospitalized children. The information
contained in these databases will augment CDER's ability to determine the
public health significance of the reports it receives through AERS, published
information, and other data sources. The acquisition of these new information
resources was made possible by awarding contracts to AdvancePCS, Premier, Inc,
and CHCA.
The AdvancePCS database will allow
FDA to examine how long non-hospitalized patients stay on prescription
medication therapy and to learn drug combinations that may be prescribed to
the same patients at the same time. The contract awarded to Premier Inc., a
strategic alliance of more than 200 hospitals and healthcare systems
nationwide, will provide similar information on hospitalized patients. The
CHCA database contains information on the use of medicines in hospitalized
children. This information will support the ongoing initiative at FDA to
approve more prescription drugs for use in children.
Contact Person: Judy Staffa, Ph.D.,
R.Ph. 301-827-3178
Back to Top
General Practice Research Database
(GPRD)
Initiative
The UK's GPRD is the largest
pharmacoepidemiologic database in the world with the highest quality data. The
database covers about 3 million lives with data going back 10 years. The data
are collected from the computerized medical record systems of 5% of all GPs in
the UK.
This database resource is superior in
many ways to any US-based database known to OPDRA. Having in-house access
would permit a wide range of crude, "fast and dirty" safety studies
as well as a wider variety of in-depth studies of interest to CDER to be
performed. In-house access would remove the constraint of working through
grantees and provide a basis for training and retaining epidemiology staff as
well as attracting new talent.
Contact person: David J. Graham,
M.D., 301-827-3238
IX.
International Activities
International Conference on
Harmonization (ICH)
Members of OPDRA have played a major
role in the ICH process. Among the successes of the past fiscal year include
Step 4 signoff of the E2B maintenance document at ICH V in San Diego. This was
a necessary step to successful implementation of international electronic
transmission of adverse event reports and was a major stumbling block until
the issues regarding unique identifier, time stamps and causality matrix were
worked out by the ICH E2B expert working group. OPDRA members also have
participated in the continuing development and maintenance of the Medical
Dictionary for Regulatory Activities (MedDRA). CDER is represented by Dr.
Honig on the MedDRA Management Board and Drs. Toni Piazza-Hepp, Sally Singer
and Andrea Neal have played a major role in developing coding criteria for
adverse event reports using MedDRA. Currently, OPDRA is working to develop
MedDRA training for ORM medical officers.
Contact Person: Andrea Feight (Neal),
DMD, MPH 301-827-3219
Video-conferences
Two separate series of regular
video-conferences are held with counterpart regulators in other countries. One
series consists of bimonthly meetings with representatives from the Bureau of
Licensed Product Assessment of Health Canada, and the Therapeutic Goods
Administration of Australia. The second series consists of 8 video-conferences
per year with the European Agency for the Evaluation of Medicinal Product, the
EMEA, specifically the Pharmacovigilance Working Party of the Committee for
Proprietary and Medicinal Products. The meetings are generally one hour in
length and involve a variety of topics including:
· Specific ADR topics and
epidemiological investigations
· Compliance issues
· Medication errors
· Consumer reporting
· Pharmacovigilance methodology
· Regulatory policy and
procedures
· Good pharmacovigilance
initiatives
· Periodic safety update
reporting experiences.
The utility of these communications
is becoming more broadly recognized. Confidentiality agreements are in place
with individual national regulatory authorities, which allow discussion of:
Contact: Andrea Feight (Neal), DMD,
MPH, 301-827-3219
International Visitors
OPDRA also hosts international
visitors on a regular basis. Since April 2000, OPDRA has hosted regulators
from Canada, Australia, New Zealand, Japan, Singapore, Taiwan, Sweden, South
Africa and Armenia as well as the World Health Organization. This has resulted
in one Foreign Regulator Series presentation (Australia and New Zealand).
The building of these relationships
has had benefits as EMEA, Canada and Japan have given us advance notice of
impending regulatory actions thus allowing FDA to prepare for industry, press
and public inquiries.
Contact: Andrea Feight (Neal), DMD,
MPH 301-827-3219
AERS to Canada
There is progress to report on the
initiative to share the AERS application with our postmarketing regulatory
counterparts at Health Canada. A draft memorandum of understanding has been
shared and negotiations on technical requirements are underway. A recent
videoconference has paved the way to successful implementation of a shared
data resource between the two countries.
Contact: Andrea Feight (Neal), DMD,
MPH 301-827-3219
In FY 2002, activities described
above will continue. In order to support the ODS reorganization, two processes
will be formalized:
· managing the Vigimed listserv
information exchange
· finding more efficient ways to
collaborate with CDER review division staff to share international
postmarketing safety surveillance information.
OPDRA also intends to expand the
amount of information available from foreign regulators on its intranet web page. OPDRA also hopes to make resources available to foreign regulators,
such as web-based materials or videotaped educational presentations, that
would obviate the need for on-site visits.
Back to Top
X. Patient Safety Initiatives
QuIC responses to IOM report
In February 2001, the FDA, with
significant input from CDER and OPDRA, released a report entitled "Final
Summary of Food and Drug Administration (FDA) Action Items - Doing What Counts
for Patient Safety: Federal Actions to Reduce Medical Errors and Their
Impact" (http://www.fda.gov/cder/drug/MedErrors/patientsafety.htm).
The report specifically outlines the steps that FDA has taken in response to
action items in the February 2000 Report to the President from the Quality
Interagency Coordination Task Force (QuIC) entitled "Doing What Counts
for Patient Safety: Federal Actions to Reduce Medical Errors and Their
Impact." OPDRA played a significant role in accomplishing a number of the
action items listed in the QuIC report.
The first action item was identified
as the need to hold a National Summit on Drug Safety. The concept recognizes
that drug safety is a complex issue that requires input from all stakeholders
and interested parties. In response, many outreach initiatives in which OPDRA
played a major role were conducted on issues such as barcoding, safety
reporting, and other aspects of drug safety. For example:
· On October 5 and 6, 2000, FDA
participated in a major drug safety think tank sponsored by Wake Forest
University in Winston-Salem, North Carolina. This meeting brought together
thought leaders from academia, industry and FDA to discuss the challenges
in minimizing the occurrence of patient injury due to pharmaceuticals.
· FDA worked with the Centers for
Education and Research in Therapeutics (CERTs) to develop a workshop on
risk communication which took place on April 30-May 1, 2001. This meeting
assembled senior representatives from FDA, the Pharmaceutical Research and
Manufacturers of America (PhRMA), and experts in the field of risk
communication. This workshop examined the challenges of changing
inappropriate prescribing behavior through communication, and set a
research agenda that is currently being pursued.
· Another action item is a
"Report to the Public on the Safety of Drugs, Devices, and
Biologics". FDA has always been committed to providing the public
with timely safety information, and to further this effort, OPDRA
developed a new Medication Errors web page (
http://www.fda.gov/cder/drug/MedErrors/default.htm).
The new web page provides information about medication error monitoring,
drug products associated with medication errors, and other resources for
finding out about medication errors.
OPDRA significantly helped in
responding to additional action items including:
· Initiating programs to develop
additional standards for drug names
· Initiating development of
packaging standards to prevent dosing and drug mix-ups
· Developing new label standards
for drugs to address errors related to medications
· Intensifying efforts to ensure
manufacturers comply with standards
· Providing access to databases
linked to healthcare systems
· Completing online adverse drug
event reporting system
· Strengthening FDA's analytic and
investigative capacity
· Strengthening FDA's outreach
activities and collaboration with federal agencies
Back to Top
Patient Safety Task Force
As a direct outgrowth of the IOM
report "To Err is Human" and the QuIC response discussed above, a
multi-agency Patient Safety Task Force was organized. The task force includes
AHRQ, CDC, FDA and HCFA. The task force was developed by the Department of
Health and Human Services to integrate the collection of data on medical
errors and adverse events, to coordinate research and analysis efforts, and to
collaborate on reducing the occurrence of injuries that result from medical
errors. Drs. Honig, Himmel, and Phillips participate in the Patient Safety
Task Force and are working to ensure that these worthwhile initiatives are
carried out in a manner, which is compatible to both the FDA and CDER mission.
At present, a major initiative by the
task force has been the development of a common reporting portal that can be
used by the healthcare community to report adverse events and errors and would
streamline the current triage process. An additional long-term goal is the
potential for sharing of safety information across the involved Agencies.
These initiatives raise numerous complex issues for FDA, such as
de-identification of reports, need for adequate information on the nature of
the event, access to the data, etc.
In parallel, a patient safety working
group was established at FDA by Dr. Janet Woodcock, which has representation
from CDER, CBER, CDRH and OC. This working group coordinates FDA's position on
safety issues across its Centers to ensure good representation of FDA's
concerns and issues at the HHS task force. OPDRA participates on this working
group as well.
As noted above, the issues
surrounding patient safety are complex, and to encourage involvement on the
part of all stakeholders, a national summit on patient safety was held this
past spring in Washington DC. The keynote speaker at the event was HHS
Secretary Tommy Thompson. The summit was organized around four major themes:
· Participation of Federal, State
and local governments in health data collection
· Confidentiality and privacy of
health data
· Maximize quality and usefulness of
data while minimizing collection burden
· Public health data collection as a
local issue
OPDRA played a key role in helping to
organize this summit, along with fellow FDA staff and staff from the other
three participating agencies. In addition, Dr. Honig co-authored an issue
paper entitled "The Importance of Definitions for Patient Safety
Information Systems" and Dr. Himmel co-authored a paper on Active vs.
Passive Surveillance.
Back to Top
Other Safety Initiatives
In addition to the continuing work
with the Patient Safety Task Force, OPDRA has helped support safety
initiatives undertaken by the National Patient Safety Foundation, the National
Coordinating Council for Medication Errors Reporting and Prevention, which is
currently chaired by Jerry Phillips, and the Medication Error Task Force,
which is a subcommittee of the QuIC.
Finally, over the past year, Temple
University School of Pharmacy, the Institute for Safe Medication Practices,
and FDA announced the nation's first certificate program in medication safety
as part of Temple's Doctor of Pharmacy program. The Temple Pharmacy 12-credit
educational track will include coursework in pharmacoepidemiology, risk
management, medication error prevention, safe medical product design, and
adverse drug reaction recognition, surveillance and prevention. Members of the
OPDRA medication errors staff have been involved in developing this program,
as well as lecturing at Temple University.
Back to Top
XI. Risk Management
Introduction
In recent years, the term risk
management has been used widely to mean many different things, depending on
one's perspective and the types of risk being addressed. From OPDRA's
perspective, risk management is the activity that results in minimizing the
risks of adverse events and medications errors associated with the use of
drugs. This includes, for example, product labeling, development of drug
distribution systems, registries, etc. At the present time, the tools that are
available to the Agency, based on our regulatory authority include labeling,
healthcare provider letters, restricted distribution, subpart H and drug
withdrawal. The Center may also issue Public Health Advisories and other
communications to the public about specific issues. The ability of some of
these tools to affect change is limited. In addition, at present, standardized
approaches as to which tools to use, when and in what combination, how their
effectiveness is assessed and other similar issues are lacking. OPDRA has
recognized the need to address these concerns and be better informed regarding
the gamut of issues surrounding risk management and has undertaken a number of
initiatives over the past year.
Risk Management Toolbox
The risk management toolbox is an
effort to systematize the approach to the risk management of drugs. Such an
effort involves an evaluation of all the components of the healthcare delivery
system, including physician, pharmacist, patient, drug company and others, to
determine where processes can be put into place to appropriately manage
certain risks. Clear objectives need to be described, processes must be
developed and criteria for invoking such systems must be established. We
recognize, however, that to bring clarity and predictability to risk
management, a broader approach must be considered. The ultimate goal is to
develop "off the shelf" systems used consistently for all drugs that
require risk management. Discussions are just beginning between CDER and PhRMA to
determine what steps are necessary to advance this effort. It is envisioned
that this will be a multi-year initiative involving broad input from experts,
stakeholders, advisory committees and other affected groups.
Back to Top
Advisory Committee Support
Drug safety, risk management, risk
communications, medication errors and patient safety are all issues which have
gained prominence over the past few years. In part, this is due to publication
of the Institute of Medicine report "To Err is Human" as well as a
greater awareness that medications are not safe in an absolute sense. The
safety of medications depends on the manner in which the healthcare community
uses them. These issues play a significant role in CDER's overall evaluation
of the risk benefit of drugs, and are common topics of discussion at advisory
committee meetings.
In general, however, these advisory
committee meetings tend to be comprised of disease/medical subspecialty
experts, rather than experts in risk related issues. At best, we have been
able to supplement some meetings with risk management experts in an ad hoc
manner. Such an approach does a disservice to the importance and complexity of
the issues. Because of these concerns, as well as OPDRA's need to obtain
scientific input on a broad range of issues, a working group in OPDRA has been
designing a drug safety advisory and risk management committee. This committee
will include experts in the areas of medication errors, risk communication,
risk perception, risk management, clinical trial methodology, evidence based
medicine, biometrics and pharmacoepidemiology. At present, this committee has
been developed as a subcommittee to an already standing advisory committee;
the Advisory Committee for Pharmaceutical Science, however, because of the
importance of this effort, our long-term plan is to ultimately make this a
standing advisory committee. It is anticipated that the committee will be up
and running in early 2002.
Contact Persons: Martin Himmel, M.D.
- 301-827-3219
Kathleen Bongiovanni - 301-827-3191
Back to Top
Risk Management White Papers
In addition to gaining input from
outside experts, it is also important to internally consider the extent of our
knowledge base and the future direction of our efforts. Two such initiatives
have taken place over the past year. Early this year, a CDER working group,
directed by Mary Willy, developed a white paper on active surveillance. This
document provides an extensive catalog of active surveillance initiatives
throughout the world and describes the advantages and disadvantages of various
approaches. The document closes with a discussion of how active surveillance
could have impacted on the ultimate removal of a number of drugs over the past
decade and suggestions for future pilot projects.
A second effort underway, in
collaboration with CDER staff, is the development of a risk management white
paper. This white paper will define the problem, describe a viable approach to
resolving the problem and develop future objectives. Dr. Peter Honig is the
OPDRA co-author for this initiative.
Drug Safety Seminar Series
Earlier this year, during the process
of developing our advisory sub-committee and working on a number of drug
specific risk management programs, it became apparent that OPDRA staff and all
of CDER would benefit from having direct contact and input from others who are
also grappling with these issues. We believe that academicians, practitioners,
researchers and trade groups may have information that could help us design
better programs and that keeping the lines of communication open would be
important for the future. To that end, a drug safety seminar series was
developed in order to bring in leaders from those areas that could impact on
risk management. Some examples of recent seminars include:
The Pharmacist's Role in Risk
Management: Susan Winckler RPh, JD - American
Pharmaceutical Association
Changing Prescribers' Behavior:
Peter Gross, MD - Hackensack
University Medical Center
Review of the Dofetilide Risk
Management Program: Nancy Allen-LaPointe, Pharm.D.
& Judy Kramer, MD - Duke CERT
Privacy and Health Information:
Potential Impact on Risk Management Initiatives, James Hodge JD - Project
Director, Center for Law and Public Health, Georgetown University
OPDRA welcomes feedback on this
series as well as suggestions for future topics and speakers.
Contact Person: Kathleen Bongiovanni
- 301-827-3191
Back to Top
Fellowship Program
OPDRA recognizes that maintaining
one's education is an important link and is necessary to better protect the
public health, particularly as it relates to drug safety. OPDRA strives to
ensure that there are adequate educational opportunities for our staff, and
those who work in other parts of our healthcare system. OPDRA has taken the
lead this year on administering a CDER fellowship program for physicians.
Specifically, the Center is sponsoring a one- to two-year fellowship, designed
to identify and train qualified candidates interested in a career in
epidemiology, especially as it relates to the study of drug safety and risk.
During the program, fellows will work part time in CDER, while at the same
time, conducting course work at a local university, ultimately leading to a
Masters degree in Epidemiology /Preventive Medicine, in Public Health, or a
similar degree. The fellow will also be expected to identify and complete a
research project at FDA during their time in the program. We anticipate
enrolling our first fellow in 2002.
Contact Person: Kathleen Bongiovanni
- 301-827-3191
Back to Top
XII. Leveraging and Outreach
OPDRA understands the importance of
building strong collaborative relationships in order to improve efficiency and
productivity. Since its inception, OPDRA has worked hard to improve working
relations within CDER, other Centers, OC, other agencies and outside
government. This process sometimes involves careful negotiation and meticulous
planning, but often results in collegial and more constructive working
relationships among the scientific, regulatory and patient communities and
results in safer drug use. The following are accomplishments in the outreach
area that proved particularly significant:
Accomplishments
· The Centers for Education and
Research on Therapeutics (CERTs) represent an unprecedented opportunity for
FDA to advance its role and influence in the use of marketed medical products.
The CERTs were established under Section 409 of the Food and Drug
Administration Modernization Act. Dr. Peter Honig serves as the FDA
representative to the CERTs steering committee and has been successful in
fulfilling the goal of having the CERTs serve as a field laboratory for FDA
issues. CERTs is an Agency for Healthcare Research and Quality (AHRQ) program
whose mission is to conduct research and provide education that will advance
the optimal use of therapeutics (drugs, medical devices, and biological
products). In particular, the CERTs program seeks to increase awareness of the
benefits and risks of new, existing, or combined uses of therapeutics, and
thereby improve the effectiveness and safety of their use. Seven centers, a
coordinating center, a steering committee, and numerous partnerships with
public and private organizations make up the CERTs program. The seven centers
each focus on therapies for a particular patient population or therapeutic
area.
· Duke University - therapies for
disorders of the heart and blood vessels
· HMO Research Network -
usefulness of HMOs for studying drug use, safety, and effectiveness
· University of Alabama at
Birmingham - therapies for disorders of the joints and bones
· University of Arizona (formerly
at Georgetown) - drug interactions, particularly in women
· University of North Carolina at
Chapel Hill (UNC) - therapies for children
· University of Pennsylvania -
therapies for infection
· Vanderbilt University -
prescription drug use in a Medicaid population
Back to Top
The centers have completed several
important projects during the past 2 years. For example, the UNC center
published a study showing a link between rickets in breast-fed children and a
lack of Vitamin D supplementation, especially among black infants. As a result
of this study, the North Carolina Department of Health and Human Services made
vitamin D available free to breast-feeding women through its Women, Infants,
and Children program (WIC). Another example is the Web-based registry
developed by the University of Arizona center to collect clinical cases of
drug-induced cardiac arrhythmias (
www.qtdrugs.org). The information
collected by the registry will be used to develop 1) detailed profiles of
people most at risk for drug-induced arrhythmias and 2) a genetic test that
can identify them in advance.
The Coordinating Center helps support
the work of the centers by enhancing synergy across the centers and
disseminating information developed by the centers. Serving as counsel to the
seven centers and the Coordinating Center is a Steering Committee, whose
members include representatives from each center, the government, the private
sector, and consumer groups.
A core value of the CERTs program is
the belief that collaboration among groups with different perspectives and
resources is critical if the results are to be applicable in the "real
world." All of the centers work with public and private collaborators on
projects, which allows the centers to expand the number of projects they work
on and their impact.
At the program level, the CERTs
collaborate with other organizations interested in advancing the best use of
therapeutics. One example is the Partners to Advance Therapeutics (PATHs)
program. In March 2001, the CERTs held the first annual PATHs meeting to
provide a forum for organizations to explore potential collaborations in
improving the use of therapeutics. A second meeting is planned for March 2002.
Another collaborative project is an
expert series of workshops focusing on the risks of therapeutics. CERTs is
hosting the workshops in collaboration with AHRQ, the U.S. Food and Drug
Administration (FDA), and the Pharmaceutical Research and Manufacturers of
America (PhRMA). The goal of the Risk Series is to develop a research agenda
to improve the assessment, communication, and management of therapeutic risk.
Back to Top
Other projects include:
· In an effort to properly gauge
interest among selected hospital pharmacists regarding their interest in
participating in a medical product surveillance system, OPDRA is conducting
focus group testing using the services of a CDRH contractor. The current
system, MedSuN, currently is a reporting mechanism that collects and submits
information to CDRH on device problems. The OPDRA focus group test will be
conducted in November, 2001 and the final report will be completed by January,
2002. These data will be used in planning the design of the drug component of
MedSuN.
· Min Chen, Pharm. D. is the
designated OPDRA liaison to CFSAN. She and others evaluated adverse events
reported with the use of ephedra and St. John's Wort, and assisted in
responding to a GAO audit on the "Risk of Harm from Questionable Health
Products and Services to the Elderly".
· OPDRA staff is collaborating with
HCFA's Center for Medicare and Medicaid Services to pilot the concept of
"safety signal amplification". In this project, FDA is seeking
adverse drug event cases to supplement its own AERS reports of rare and
well-defined adverse events related to cases of liver failure associated with
Parkinson's disease treatments (Tasmar or Comtan). The amplification is among
a population of approximately 37 million elderly patients covered by Medicare.
Using a specially-developed algorithm, the Virginia Healthcare Quality Center
has identified approximately 19 cases of hospitalized liver failure among
elderly Parkinson's patients and will be making these hospital records
available to FDA to assess the potential association with either Tasmar or
Comtan. FDA will take corresponding regulatory risk management actions based
on their findings and completion of this project is anticipated by January
2002. As a result of this collaboration, the Center for Medicare and Medicaid
Services is making $250,000 available annually for its contractors to assist
FDA in addressing priority drug safety issues among the elderly by signal
amplification.
· By working with the Consumer
Products Safety Commission, OPDRA's epidemiology staff evaluated the utility
of the National Electronic Injury Surveillance System (NEISS) data to detect
patients presenting to emergency departments with specific adverse events due
to drugs. It was determined that although this system does detect some
patients with known, labeled reactions, it does not currently provide enough
detail to be of use to FDA for surveillance purposes.
· The medication errors staff with
help from CDER's Office of Training and Communications established an Internet
site on CDER's homepage. This website provides information to the public on
current medication error issues.
· The medication errors staff is
writing quarterly articles for Drug Topics that reach out to a large pharmacy
audience.
Back to Top
In addition FDA has collaborated in:
· a program to increase the
appropriate use of beta blockers in patients with congestive heart failure,
and
· a program to evaluate the
effectiveness of physician education in the launch of a new drug to treat
atrial fibrillation.
American Association for the Study of
Liver Diseases/FDA/PhRMA Hepatotoxicity Workshop
A multidisciplinary conference was
held in February 2001 to discuss drug-induced liver injury. Topics included
preclinical mechanistic studies and models for human hepatotoxicity, early
clinical signals in controlled trials, and strategies to improve postmarketing
surveillance of marketed drug products that may be potential hepatotoxins. As
a result of this conference, the Division of Over the Counter Drugs and OPDRA
evaluated the problem of acetaminophen-related hepatotoxicity in preparation
for a future public meeting. For more information, see
http://www.fda.gov/cder/livertox/.
Contact: John Senior, M.D.,
301-827-7280
Back to Top
XIII. Regulatory Research
OPDRA has engaged in research that
has served to inform CDER on policy development and appropriate regulatory
action. Specifically, we have engaged in research activities that have
attempted to characterize the use, consistency of application, and
effectiveness of CDER risk management and communication efforts.
Some selected research activities
include:
· In conjunction with the Georgetown
CERT, a survey of undergraduate and graduate medical program directors was
conducted attempting to characterize and quantify the amount of instruction on
adverse event identification and reporting. The results of this survey were
published, in part, in the Journal of the American Medical Association. It was
apparent that these topics were generally not part of the curriculum in the
undergraduate medical or residency training programs. In response to these
findings, CDER and the Georgetown CERT developed a teaching module on the
principles of clinical pharmacology and adverse reactions to drugs. This
module was presented to representatives of the American Association of Medical
Colleges (AAMC) and will be made available for incorporation into the medical
training curricula through the CERTs.
· Epidemiologists in OPDRA have been
active in developing new methodologies for assessing the benefit and risk of
drugs by applying traditional epidemiological techniques and newer modeling
methods to the analysis of clinical trial safety databases. This promising
approach will be advanced through a CDER RSR grant application in the upcoming
year.
· Zili Li and David Graham have
worked closely with the Gastrointestinal Drugs Division Staff to assess time
trends of adverse events in premarketing clinical trials submitted in the
alostron clinical trial safety database NDA.
Back to Top
XIV. Regulations, Guidances and MaPPS
OPDRA has been involved in supporting
several critical regulations and guidances that address postmarketing
reporting and patient safety. These include:
· Notification to accept e-sub
ISRs without attachments posted on May 18, 2001.
· Proposed Suspected Adverse
Reaction Rule (SADR); cleared by FDA; now in HHS.
· Draft Guidance for Industry:
Providing Regulatory Submissions in Electronic Format-Expedited Adverse Drug
Experience Reports (under development)
· Draft Guidance for Industry:
Providing Regulatory Submissions in Electronic Format-Postmarketing Periodic
Adverse Drug Experience Reports (under development)
· Draft Guidance for Industry on
Periodic Safety Update Reports (under development)
· Proposed Regulation on
Electronic Expedited Adverse Event Reporting (under development)
· Draft Guidance entitled, "Postmarketing
Safety Reporting for Human Drug and Biologic Products Including
Vaccines", published in 3/01, generated approximately 600 public
comments; comments under review
· Bar coding proposed regulation
(under development)
· Draft guidance to industry on
proposed tradenames and packaging (under development)
· Guidance for Industry:
Submitting Proprietary Names for Evaluation (under development)
· MaPP: Minimizing Medication
Errors associated with confusing drug names, labels, or packaging (under
development)
· Guidance for Industry:
Minimizing Medication Errors - Names, Labeling and Packaging (under
development)
Back to Top
Conclusion
This has been an intensely busy year
for OPDRA. OPDRA's increased workload and new responsibilities that will be
assessed through the upcoming reorganization may be attributed to our strong
belief in the importance of nurturing trust within the Center and
demonstrating value in an effort to ensure that drugs are used as safely as
possible. We look forward to maturing as an Office and moving forward with
several strategic planning initiatives.
The first priority for the new Office
will be the tactical implementation of the reorganization; including selecting
appropriate people to lead the different ODS divisions and establishing
internal procedures for already existing and newly assigned responsibilities
(e.g. MedWatch and patient labeling). We will also have to develop processes
to incorporate new data resources into the business of safety evaluation.
A second, but equally important
priority, for the new Office will be the development of ODS/ORM operating
principles. It is critical that ODS maintains close communication and
synergistic relationships with ORM and the ORM reviewing divisions.
Codification of relationships through MaPPs on such issues as tradename
reviews, postmarketing safety issues, evaluation of risk management programs
and medication guides will be addressed. Furthermore, the shared
responsibilities in the review of Periodic Safety Update Reports (PSURs) will
be addressed through a MaPP and a guidance to industry is also necessary. The
appropriate use of the new Drug Safety and Risk Management Advisory Committee
will also be codified through a MaPP. However, it will be very important for
ODS to maintain regular participation in ORM activities by attending the
critical ORM meetings such as ministaff, division director meetings, etc.
The final priority for the new office
will focus on the CDER initiative relating to Risk Management. The development
of the Risk Management White Paper, the identification of a risk management
and risk communication research agenda, the successful launch and utilization
of the new Drug Safety and Risk Management Advisory Committee, the optimal use
of MedWatch and the CERTs for risk communication and outreach, the
identification of additional data resources to optimize risk assessment, and
ultimately, the codification of ODS' role in risk management for marketed
drugs will represent major opportunities and challenges for the next year.
Back to Top
Appendix 1 - OPDRA Organizational
Chart
Current OPDRA Units

Back to Top
Appendix 2 - OPDRA Meeting Presentations
Beam S. "Update of Proposed
Proprietary Name Reviews" DIA Annual Meeting, July 2001.
Bharati M, Drinkard C, Shatin D,
Graham D. "The Risk of Esophageal Obstruction Associated with an
Anti-Allergy Medication (Claritin-D® 24 Hour)" United Health Group,
Minnetonka, MN; Food and Drug Administration, Rockville, MD. Abstract
presentation at the Academy for Health Services Research and Health Policy
annual meeting, June 2001.
Bharati M, Drinkard C, Shatin D
(presenter), Willy M, Graham D. "Cylert® Labeling Compliance: Second
Line Therapy Use and Liver Function Monitoring" Abstract presentation at
the International Society of Pharmacoepidemiology, August 2001.
Bongiovanni K. "CDER 101 -
Overview of CDER's Postmarketing Safety Evaluation", Presentation to
Pharmaceutical Industry, May, 2001.
Diwa D, Fan J, Kim H. Mahmud A.
"Medications and the FDA" National Boards of Pharmacy, October 2001.
Diwa D, Fan J, Kim H, Mahmud A.
"Reporting Medication Errors: A Public Health Responsibility" Annual
Commissioned Officers Association Meeting, May 2001.
Graham D. "CDER 101 -
Epidmiology Approaches to Postmarketing Drug Safety at CDER",
Presentation to Pharmaceutical Industry, May 2001.
Graham DJ. "Postmarketing
resources and their utilization within the Center for Drug Evaluation and
Research." Seminar presented at course in Clinical Trials, Center for
Devices and Radiologic Health, Rockville, MD, June 1, 2000.
Graham DJ. Collaboration between
academia and government agencies for study of drug safety issues. 16th
International Workshop on Postmarketing Surveillance, Saint-Paul de Vence,
France, June 27, 2000.
Graham DJ. Modeling cumulative risks
and hazard rates using postmarketing data. FDA/Industry Workshop on
Statistically Sound Decision-Making, Bethesda, MD, September 15, 2000.
Graham DJ. Data resources at FDA for
postmarketing safety evaluation and applications of their use. Center
Scientific Rounds, December 13, 2000.
Graham DJ. "Interactive problem
solving: how would you have epidemiologically studied the problem of liver
failure with troglitazone?" Presented at a course in Pharmacoepidemiology,
Yale University School of Medicine, New Haven, CT, April 10, 2001.
Graham DJ. "Epidemiologic
approaches to drug safety at FDA," presented at course sponsored by the
Drug Information Association, Washington DC, October 25, 2001.
Graham DJ. "Principles of
pharmacovigilance and methods of practice at FDA," presented to the
Nutritional Supplements Subcommittee, Institute of Medicine, Washington, DC,
November 29, 2001.
Graham DJ. "Models of excellence
in pharmacovigilance." 17th International Conference on
Pharmacoepidemiology, Toronto, Canada, August 25, 2001.
Manda B, Drinkard C, Willy E, Graham
DJ, Shatin D. Compliance with labeling for Cylert: use as second-line therapy
and liver function monitoring. 17th International Conference on
Pharmacoepidemiology, Toronto, Canada, August 24, 2001.
Himmel, M. Johns Hopkins Graduate
Training Program in Drug Development - Panel discussant of student drug
development programs, 12/2000.
Himmel, M. PERI - "PSURs: An FDA
Perspective" - 3/2001.
Himmel, M. PERI - "Postmarketing
Drug Risk Assessment Initiatives at CDER" - 3/2001.
Himmel, M. PERI - "PSURs: An FDA
Perspective" - 11/2001.
Himmel, M. Johns Hopkins Graduate
Training Program in Drug Development - Seminar on Postmarketing Drug Risk
Assessment at FDA - 12/2001.
Holquist C. "CDER 101 -
Medication Errors and FDA Perspective", Presentation to Pharmaceutical
Industry, May, 2001.
Holquist C. "CDER 101 -
Medication Errors and FDA Perspective", Presentation to Pharmaceutical
Industry, October, 2001.
Honig P. Regulatory Affairs
Professional Society (RAPS) Annual Meeting "Risk Management: Impact on
the Consumer" Washington, DC October 2000.
Honig P. Wake Forest Drug Safety Symposium" The
Adverse Event Reporting System for Drugs in the United States" Winston-Salem, NC October 2000.
Honig P. Pharmaceutical Education
& Research Institute (PERI): Adverse Events: A Review of Regulations and
Challenging Exercises in Pharmacovigilance.
Honig P. "Postmarketing Drug
Risk Assessment and Initiatives at CDER" Arlington, VA October 2000.
Honig P. European DIA Meeting,
"Periodic Safety Update Reports: US Regulatory Perspectives, London, UK
December 2000.
Honig P. FDA-PhRMA-AASLD
Hepatotoxicity Workshop "Postmarketing Perspectives: State of the Art and
Future Initiatives Chantilly, VA February 2001.
Honig P. American Society of Clinical
Pharmacology and Therapeutics "FDA Perspectives on Drug Labeling as a
Risk Management Strategy" Orlando, Fl March 2001.
Honig P. American Society of Clinical
Pharmacology and Therapeutics Chair, Public Policy Session on
"Translating Research Into Practice" Orlando, FL March 2001.
Honig P. American Society of Clinical
Pharmacology and Therapeutics Government Affairs Committee.
Honig P. "Proposed Changes to
the Professional Labeling Regulations" Orlando, FL March 2001.
Honig P. Temple University
FDA/Industry Conference, "FDA's Drug Risk Management Initiatives"
Philadelphia, PA April 2001
Honig P. DIA Pharmaceutical Outcomes
Meeting, "FDA Patient Safety Initiatives" Savannah, GA April 2001
Honig P. FDA Office of Women's Health
Dialog Session, "Dialog on Federal Patient Safety Initiatives"
Washington DC April 2001.
Honig P. Annenberg 3: Let's Talk:
Communicating Risk and Safety in Health Care, "FDA and the Risk
Management of Medical Products: Impact on the Patient", St Paul, MN, May
2001.
Honig P. PERI: Clinical Trials Course
"Regulatory Perspectives on Metabolism-Based Drug-Drug
Interactions", Washington DC, May 2001.
Honig P. International Conference on
Harmonization (ICH), Postmarketing Surveillance Workshop on Postmarketing
Surveillance, "FDA Perspectives on PSUR (E2C) Implementation",
Tokyo, May 2001.
Honig P. Infectious Disease Society
of America Symposium: Effective Strategies for Improving Patient Safety and
Preventing Medical Errors in Infectious Diseases, "FDA Actions to Reduce
Medical Errors and Their Impact", Chicago, IL July 2001.
Honig P. Regulatory Affairs
Professional Society (RAPS) Meeting "Future Perspectives on Risk
Management", Washington, DC August 2001.
Honig P. Regulatory Affairs
Professional Society (RAPS) Annual Meeting, "CDER Risk Management Tools
and their Utility" Baltimore, MD, November 2001.
Honig P. First Annual DIA Workshop in
Japan on Global Pharmacovigilance ,"Postmarketing Safety Surveillance and
Risk Management of Marketed Drugs", Tokyo, November 2001.
Honig P. First Annual DIA Workshop in
Japan on Global Pharmacovigilance "Postmarketing Safety Studies: US
Regulatory Perspective" Tokyo, November 2001.
Kweder S, Holmboe E, Shaffer D, and
Sacks L. "Managing drug risks: from policy to bedside". The Society
of General Internal Medicine 24th Annual Meeting - Workshop. San
Diego, CA, May 3, 2001.
Manda B, Drinkard C, Willy M, Graham
D, Shatin D. "Compliance with labeling for pemoline (Cylert): use as
second-line therapy and liver function monitoring". International
Conference on Pharmacoepidemiology, Toronto, Canada, August, 2001.
Neal A. Global Challenge in
Pharmacovigilance, "Exchange of Safety Information between the FDA and
other Regulators". Drug Information Association Annual Meeting, Denver,
CO, July, 2001.
Neal A. "FDA on MedDra
Implementation". MedDRA MSSO User's Group Meeting, Denver, CO, July 2001.
Neal A. "MedDRA Implementation
at the FDA: an Update". Pharmaceutical Education and Research Institute,
Arlington, VA, May 2001.
Neal A. "Lessons Learned in
Implementing MedDRA at the FDA", Pharmaceutical Education and Research
Institute, Adverse Events: A Review of Regulations and Challenging Exercises
in Pharmacovigilance, Arlington, VA, March 2001.
Neal A. "MedDRA Implementation
at the FDA: an Update", DIA Drug Safety Surveillance and Epidemiology
Training Course, Washington, D.C., April 2001.
Neal A. "Lessons Learned in
Implementing MedDRA at the FDA", DIA Drug Safety Surveillance and
Epidemiology Training Course, Washington, D.C., October 2000.
Neal A. "MedDRA Implementation
and the FDA: an Update". Pharmaceutical Education and Research Institute,
MedDRA: An Update, Philadelphia, PA, December 2000.
Neal A. "MedDRA Implementation
for Regulatory Reporting". MedDRA MSSO User's Group Meeting, San Diego,
CA, November 2000.
Nourjah P. "The Value of NEISS
Data as a Resource for Postmarketing Surveillance of Drug Products",
Interagency Meeting, Bethesda, Maryland, August 14, 2001.
Piazza-Hepp, T. Postmarketing Safety
Evaluation, "CDER 101" Course, cosponsored by DIA/CDER Staff
College, Washington, D.C., May 2001.
Piazza-Hepp, T. Overview of AERS and
Postmarketing Safety, to Vitali Pool, M.D., CDC Epidemiologist, VAERS Project,
June 2001.
Piazza-Hepp, T. MedDRA: Current
Issues, to Clinical Safety Committee of PhRMA, Washington, D.C., September
2001.
Senior J. "Surveillance for
Hepatotoxicity", National Institutes of Health workshop
on Mechanisms and Test Systems of Drug-Induced Liver Injury,
Lister Hill Center, Bethesda MD, October 2000.
Senior J. "Drug Development and
review at the FDA", Science for Physicians
Lecture Series, University of Texas Southwestern, Dallas TX, February 2001.
Senior J. "Overview of the
problem of drug-induced liver injury, at session on Drug-Induced Liver
Injury: A Global Problem", 27th Annual Summer Meeting of the Toxicology
Forum, Given Institute, Aspen, CO, July 2001.
Senior J. "Data sources and
descriptors of hepatotoxicity, at special session on "Biomarkers of
Toxicity", Toxicology Discussion Group, 7th Annual Meeting of the Society
for Biomolecular Screening, Baltimore Convention Center, September 2001.
Shaffer D. "QT Prolongation and
torsade de pointes: From idiosyncrasy to postmarketing analyses".
International Symposium on Mechanisms, Models, and Predictions of
Idiosyncratic Drug Toxicity. Atlantic City, NJ, June 11-13, 2001.
Shaffer D, Singer S, Korvick J.
"Macrolide and fluoroquinolone associated torsade de pointes: review of
the FDA Adverse Event Reporting System". The 41st Annual
Interscience Conference on Antimicrobial Agents and Chemotherapy. Chicago, IL,
September 22-25, 2001. (Rescheduled to December 16-19 2001.)
Toyer D. "Sildenafil and
hemorrhagic events", COA Public Health Professional Conference, Alexandria, A, May 2001.
Trontell, Anne. "Evaluating the
Impact of Risk Management Programs - OPDRA Capabilities &
Experience", CDER CASE Seminar November 2000.
Trontell Anne. "CDER Initiatives
in PostMarketing Drug Risk Assessment", DIA Conference on
Pharmacovigilance in the New Millennium, January 2001.
Trontell Anne. "CDER's
Postmarketing Safety Program", 2 presentations at DIA Drug Safety and
Surveillance Course, April and October 2001.
Trontell Anne. "Adverse Drug
Events/Reactions: FDA Definitions and Databases",
NIH Conference on Pediatric Adverse Drug Reactions, April 2001.
Trontell Anne. "Developing Risk
Management Programs Post-Approval: Options, Experience, and Lessons
Learned", DIA Annual Meeting, Plenary Session, July 2001.
Trontell Anne. "Update of OPDRA
Postmarketing Activities, DIA Annual Meeting", OPDRA Update Session, July
2001.
Trontell Anne. "Risk
Communication: FDA Perspectives and Initiatives International Society for
Pharmacoepidemiology", Plenary Session on Risk Communication, August
2001.
Trontell Anne. "CDER's
Postmarketing Safety Program" (2 presentations at DIA Drug Safety and
Surveillance Course, April and October 2001).
Trontell Anne. "DDRE1 Safety
Surveillance and Risk Management" (2 presentations, to the Divisions of
Oncologic Drug Products and the Office of Clinical Pharmacology and
Biotherapeutics) .
Trontell Anne. "CDER's
Postmarketing Safety Program" , New Reviewer's Workshop, April 2001.
Uhl K. An FDA perspective. Gordon
Research Conference on Medicinal Chemistry; "Drug Safety in the 21st
Century", August 2001.
Uhl K. "CDER safety surveillance
and risk management: OPDRA capabilities and experience" American Association of Pharmaceutical Scientists Indianapolis/Cincinnati Discussion
Group (I/CDG), 1st Annual Symposium on Current Topics in Drug
Development, May 2001.
Wilson J. "Electronic Submission
of Postmarketing ADR Reports", Presentation to Pharmaceutical Industry,
May 2001.
Appendix 3 - OPDRA Publications
Ahmad SR, Singer S, Leissa BG.
Congestive heart failure associated with itraconazole. Lancet 2001;
357:1766-7.
Beitz J. Trial endpoints for drug
approval in oncology: Chemoprevention. Urology 2001: 57(4A)213-215.
Brinker AD, Bonnel RA, Feight AG,
Nourjah P. Celecoxib and rofecoxib. JADA 2001;132:1497-1498.
Bross PF, Beitz J, Chen G., Chen XH,
Duffy E, Kieffer L, Roy S, Sridhara R, Rahman A, Williams G, Pazdur R.
Approval summary: Gemtuzumab ozogomicin in relapsed acute myeloid leukemia.
Clin Cancer Res 2001;7:1490-1496.
Chiao J, Beitz J, DeLap RJ.
Antimetabolic agents in Current Cancer Therapeutics, 4th
Edition, JM Kirkwood, MT Lotze, JM Yasko eds., Philadelphia 2001.
Drinkard C. MPH, PhD, Shatin D. PhD,
Clouse J. RPh, MS. "Postmarketing Surveillance of Medications and
Pregnancy Outcomes: Clarithromycin and Birth Malformations" Pharmacoepidemiology
and Drug Safety 2000:9:549-556 Authors acknowledged contribution of David
Graham MD, MPH at the Food and Drug Administration.
Fanning MM, Wassel R, Piazza-Hepp T.
Pyrogenic reactions to gentamicin therapy. N Engl J Med 2000; 343:1658-9.
Galson S, Kweder S, Houn F,
Raczkowski V, Honig P. The FDA and the Lancet: an exchange. Lancet
2001; 358:415.
Gardner J, Blough D, Drinkard C,
Shatin D, Anderson G, Graham D, Alderfer R. "Tramadol and Seizures: A
Surveillance Study in a Managed Care Population" Pharmacotherapy
2000;20(12):1423-1432.
Graham D, Drinkard, C, Shatin D,
Tsong Y, Burgess M. "Liver Enzyme Monitoring in Patients Treated with
Troglitazone" JAMA 2001; 286:831-833.
Rodriguez EM, Staffa JA, Graham DJ.
The role of databases in drug postmarketing surveillance. Pharmacoepidemiol
Drug Safety 2001; 10:407-10.
Graham DJ, Waller PC, Kurz X. A view
from regulatory agencies. In: Strom BL, ed. Pharmacoepidemiology (3rd
edition). John Wiley & Sons, Ltd., New York, 2000:109-124.
Holquist C., Phillips J. "FDA
Advise-Err: Dispensing Errors Associated with Zantac and Zyrtec" ISMP
Medication Safety Alert, November 1, 2000.
Holquist C., Phillips J. "FDA
Advise-err: Medication Errors Associated with Confusion between Flomax and
Volmax" ISMP Medication Safety Alert, November 15, 2000.
Holquist C., Phillips J.,
"Safety Briefs: Antibacterial Gel or Risperdal Oral Solution?" ISMP
Medication Safety Alert, June 27, 2001.
Holquist C., Phillips J., "FDA
Safety Page: Curbing Med Errors Involving Anzemet, Bentyl" Drug Topics,
July 2, 2001.
Honig PK, Phillips J, Woodcock J. How
many deaths are due to medical errors? JAMA 2000; 284:2187-8.
Honig PK Commentary: On-demand use of
B2-agonists led to better asthma control than did regular use in
moderate-to-severe asthma. ACP Journal Club 2001;134:17.
Honig PK Commentary: On-demand use of
B2-agonists led to better asthma control than did regular use in
moderate-to-severe asthma. Evidence Based Medicine 2001;6:15.
Honig PK. Commentary: Herbal
preparations may improve FEV1 and symptoms in asthma. ACP Journal Club
2001;134:96.
Huang S-M, Honig P, Lesko LJ, Temple
R, Williams RL. An Integrated Approach to Drug-Drug Interactions- A Regulatory
Perspective, Chapter 18 in Drug-Drug Interactions: From Basic
Pharmacokinetic Concepts to Marketing Issues, Ed. A.D. Rodrigues, Marcel
Dekker, New York 2001.
Kleiner DE, Lee WH, Senior JR,
Balistreri WF. The burden of hepatotoxicity, in Bissell
DM, Gregory JG, Laskin DL, Hoofnagle JH, eds., Drug-induced liver injury:
mechanisms and test systems. [meeting report] Hepatology 2001 (April);
33:1009-13.
LeGrenade L, Myocarditis and
cardiomyopathy associated with clozapine use in the United States. NEJM, Vol
345, No 3, July 19, 2001.
La Grenade L, Graham D, Trontell A.,
Myocarditis and cardiomyopathy associated with clozapine use in the United
States. N Engl J Med 2001; 345(3): 224.
Levy D, Stergachis A, McFarland L,
Van Vorst K, Graham D, Johnson E, Park B, Shatin D, Clouse J, Elmer G.
"Antibiotics and Clostridium difficile Diarrhea in the Ambulatory
Setting" Clinical Therapeutics 2000:22(1):91-102.
Malozowski S, Green L. Reply to the
Editor. Reports of premature thelarche in girls. J. Pediatr 2001; 138(3):449.
Park B, Clouse J, Shatin D,
Stergachis A. "Incidence of Adverse Oesophageal and Gastric Events in
Alendronate Users" Pharmacoepidemiology and Drug Safety 9 371-376
(2000) Authors acknowledged the assistance of Diane Wysowski PhD, OPDRA for
initiating the study and reviewing and commenting on the results.
Phillips J, Beam S, Brinker A,
Holquist C, Honig P, Lee LY, Pamer C. Retrospective analysis of mortalities
associated with medication errors. Am J Health-Syst Pharm, October 2001;
58:1835-41.
Ray W, Meredith S, Thapa P, Meador K,
Hall K, Murray K "Antipsychotics and the Risk of Sudden Cardiac
Death" Arch Gen Psychiat In Press.
Rosebraugh CJ, Honig PK, Yasuda et
al. Formal education about medication errors in internal medicine clerkships.
JAMA 2001;286:1019.
Smalley W, Shatin D, Wysowski DK,
Gurwitz J, Andrade SE, Goodman M, Chan KA, Platt R, Schech SC, Ray W.
Contraindicated use of cisapride: impact of Food and Drug Administration
regulatory action. JAMA 2000;284:3036-3039.
Senior J. Drug-induced liver injury:
A national and global problem, internet publication of 3 "white papers" on
preclinical, clinical, and postmarketing considerations, and 11
invited presentations at www.fda.gov/cder/livertox,
posted April 2001.
Singer SJ, Tiernan R, Sullivan EU.
Interstitial pneumonitis associated with sirolimus therapy in renal-transplant
recipients [letter]. N Engl J Med 2000;343:1815-6.
Smalley W, Shatin D, Wysowski DK,
Gurwitz J, Andrade SE, Goodman M, Chan KA, Platt R, Schech SC, Ray W.
Contraindicated use of cisapride: impact of Food and Drug Administration
regulatory action. JAMA 2000;284:3036-3039.
Smalley W, Shatin D, Wysowski D,
Gurwitz J, Andrade S, Goodman M, Chan K.A, Schech S, Ray W.
"Contraindicated Use of Cisapride: Impact of Food and Drug Administration
Regulatory Action" JAMA 2000(April); 20(7):410-416.
Thomas M., Holquist C., Phillips J.,
"Med Error Reports to the Food and Drug Administration Show a Mixed Bag ,
Drug Topics, October 2001.
Trontell AE, Honig PK. Clopidogrel
and Thrombotic Thrombocytopenia Purpura. New England Journal of Medicine
2000;343:1191-92.
Trontell, A., How the US Food and
Drug Administration defines and detects adverse drug events, Current Therap.
Res. 62(9):641-649, 2001.
Uhl K, Honig PK. Risk Management of
Marketed Drugs: FDA and the interface with the practice of medicine,
Pharmacoepidemiology and Drug Safety 2001;10:1-4.
Weller PF, Plaut M, Taggart V,
Trontell A., The relationship of asthma therapy and Churg-Strauss syndrome:
NIH workshop summary report. J Allergy Clin Immunol, 108 (2): 175-183.
Wysowski DK, Corken A, Gallo-Torres
H, Talarico L, Rodriguez EM. Postmarketing reports of QT prolongation and
ventricular arrhythmia in association with cisapride and Food and Drug
Administration regulatory actions. Am J Gastroenterol 2001;96:1698-1703.
Wysowski DK, Pitts M, Beitz J.
Depression and suicide in patients treated with isotretinoin. New Engl J Med
2001;344:460.
Wysowski DK, Beitz J. Methodological
limitations of the study "Isotretinoin use and risk of depression,
psychotic symptoms, suicide, and attempted suicide," Arch Dermatol
2001;137:1102.
Wysowski DK, Pitts M, Beitz J. An
analysis of reports of depression and suicide in patients treated with
isotretinoin. J Am Acad Dermatol 2001;45:515-519.
Back to Top
Appendix 4 - Posters
Ahmad SR, Nourjah P, Trontell A. Drug
use in pregnancy. Poster presentation at ISPE, Toronto, Canada, August 2001.
Published in Pharmacoepidemiology and Drug Safety 2001; 10:S83-S84.
Ahmad SR, Trontell A, Pitts M,
Karwoski C, Beitz J. Risk of liver failure in association with antifungal
drugs for onychomycosis. Poster presentation at ISPE, Toronto, Canada, August
2001. Published in Pharmacoepidemiology and Drug Safety 2001; 10:S68.
Brinker A, Beitz J. Analysis of
spontaneous reports data for possible beneficial hypotheses. American College
of Clinical Pharmacology and Therapeutics Annual Meeting, March 2001.
Bonnel RA, Villalba ML, Midthun K,
Karwoski C. Deaths associated with inappropriate intravenous colchicine
administration. American College of Rheumatology Annual Meeting, San
Francisco, November 12-15, 2001.
Boxwell D, Toerner J. Fatal
hepatotoxicity associated with combination hydroxyurea and nucleoside reverse
transcriptase inhibitors (NRTIs): Cases from the FDA Adverse Event Reporting
System (AERS). 8th Annual Conference on Retroviruses and
Opportunistic Infections, Chicago, Illinois, February 4-8, 2001.
Brown SL, Kedas A, Bascanyi JT,
Purvis-Wynn SL, Fatal and serious complications associated with cosmetic
suction lipectomy. 17th International Conference on Pharmacoepidemiology,
Toronto, Canada, August 2001.
Green L, Zawadzki J. Thiazoline-associated
hepatoxicity: Comparison of spontaneously reported postmarketing cases in
association with troglitazone, rosiglitazone, and pioglitazone. Annual
Endocrine Society Meeting, Denver, CO, June 2001.
La Grenade L, Graham D, Trontell A,
Beitz J, Sherman R, Nourjah P. Postmarketing drug risk assessment: the
phenylpropanolamine experience. 2001 FDA Science Forum, Washington DC,
February 15-16.
La Grenade L, Estimating public
health impact of adverse drug events in pharmacoepidemiology:
phenylpropanolamine and hemorrhagic stroke. ISPE, 17th Annual International
Conference on PharmacoepidemiologyToronto, Canada, August 2001.
La Grenade L, Myocarditis and
cardiomyopathy associated with clozapine use: the US experience. ISPE, 17th
Annual International Conference on PharmacoepidemiologyToronto, Canada, August
2001.
McCloskey CA, Staffa JA. Pediatric
inpatient use of eleven selected drugs: Results of two pilot studies. 2001 FDA
Science Forum, Washington DC, February 15-16.
McCloskey CA, Staffa JA. Pediatric
inpatient drug exposures: Results of two pilot studies of eleven selected
drugs. International Society of Pharmacoepidemiology Annual Meeting, Toronto,
Canada, August 2001.
Nourjah P, Trontell A., Song Y., La
Grenade L., Graham D., Beitz J. Sparse-data bias in pharmacoepidemiology: the
Hemorrhagic Stroke Project (HSP) case-control study on PPA and risk of
hemorrhagic stroke (HS). ISPE Meeting, Toronto, Canada, August 2001.
Willy ME, Li Z. A study of product
labeling - retrospective analysis of the content of the product labeling of
prescription drugs with the risk of hepatic failure. International Conference
on Pharmacoepidemiology Toronto, Canada, August 2001.
Back to Top
1 Data
collected as of September 24, 2001
2 An
individual, usually a health care practitioner, notifies us directly of a
suspected serious adverse event.
3 These
report serious and unexpected adverse events to us as soon as possible within
15 days of discovering the problem.
4 These
report all other adverse events, such as those that are less than serious or
described in the labeling.
5 An ICSR is
an individual case safety report, also known as one adverse event report. When
an ICSR is loaded into AERS, it
is known as an ISR.
 Back to Top Back to Top  Back to About CDER
Back to About CDER
FDA/Center for Drug Evaluation and Research
Last Updated: January 25, 2002
Originator: OTCOM/ODS
HTML by SJW |


