
Panel Discussions and Recommendations
FDA Recommendations: Strategies for Assessing Gender Effects
We would like to express our gratitude to the planners of the Gender Studies
in Product Development: Scientific Issues and Approaches Workshop including:
Dr. Elaine Esber, Ms. Mary Gross, Dr. Tom Ludden, Dr. Stella Machado, Ms. Theresa
McGovern, Dr. Kimber Richter, Dr. Linda Sherman, Dr. Sol Sobel, Dr. Deborah
Smith, Ms. Terry Toigo and Dr. Laura Kragie. Some of the core planning group
also participated in the workshop as speakers or panelists including: Dr. Elaine
Esber, Dr. Kimber Richter, Dr. Laura Kragie, Dr. Stella Machado, Ms. Terry McGovern,
Dr. Sol Sobel, and Dr. Carol Trapnell.
We would also like to acknowledge speakers who participated in the workshop,
including Dr. David A. Kessler, Dr. Ruth Merkatz, Dr. Janet Woodcock, Dr. Roger
Williams, Dr. Robert J. Temple, Dr. Eugene G. Hayunga, Dr. Janice K. Bush, and
Dr. Jeanne DeJoseph. Other workshop speakers whose contributions should be acknowledged
include Dr. Janice B. Schwartz, Dr. Mei-Ling Chen, Dr. Raymond B. Woosley, Dr.
Lewis B. Sheiner, Dr. Edward E. Wallach, Dr. David A. Flockhart, Dr. Paresh
Dandona, Dr. Jean Hamilton, Dr. Scott Lucas, Dr. Richard Simon, Dr. Robert O'Neill,
Dr. Jean-Louis Steimer, Ms. Sarah Kogut and Dr. Julie Swain. Our thanks also
to the panelists at the workshop: Dr. Joy Cavagnaro, Dr. Neal Cutler, Dr. Victoria
Hale and Dr. Louis Cantilena. Special recognition is given to panel moderators:
Dr. Alan Sedman, Dr. Jean R Rowan and Dr. Murray Lumpkin. All of those mentioned
previously also contributed greatly to this summary of the workshop. Special
mention should also be given to Dr. Shiew Mei Huang, Dr. Mei-Ling Chen, Dr.
Stella Machado, Ms. Bonnie Malkin and Dr. Robert Temple for special contributions
made to the executive summary.
We would like to give special recognition of Ms. Claudette Bain who unfortunately
died from AIDS complications earlier this year. Ms. Bain offered a stirring
first-hand account from the patient's point of view that added much to the workshop
discussion and her courage will long be remembered.
On November 6 and 7, 1995, the Food and Drug Administration's Office of Women's Health, in collaboration with the FDA's Center for Drug Evaluation and Research, the Center for Biologics Evaluation and Research, and the Center for Devices and Radiological Health, convened a public workshop entitled "Gender Studies in Product Development: Scientific Issues and Approaches." The meeting was designed to explore the science involved with assessing gender effects during the development of medical products, including drugs, biologics, and medical devices, and to identify significant areas for further research and policy development.
The Gender Workshop was organized around three broad topics that affect the evaluation of medical products used by women: 1) pharmacokinetics and pharmacodynamics; 2) hormonal influences; and 3) study design and analysis.
Four critical questions were explored:
Based on the workshop conclusions and recommendations, and coupled with the 1993 gender guideline, which emphasizes enrolling adequate numbers of women in clinical trials and conducting appropriate analyses, FDA believes that a reasonably coherent picture is emerging of how and when to assess gender effects during the drug development process. Methodologies available to assess gender and other population subset effects include: 1) in-vitro studies in human and non-human tissue and in-vivo studies in non-human species; 2) early phase exploratory clinical studies using pharmacokinetic and pharmacodynamic approaches, coupled with a mechanistic understanding of drug action, if available; and 3) late confirmatory studies in humans. It is the FDA's judgment that sponsors can and should use early studies to identify the presence or absence of important gender differences and to adjust later trials to evaluate the clinical importance of these differences. Late phase studies may signal by-gender safety and efficacy differentials via post-hoc subset analyses.
This Executive Summary captures highlights of the two-day workshop and places the issue of identifying gender-related drug effects in an historical context. The discussions and recommendations of the three panels are summarized here; detailed excerpts from the speaker presentations are available in a separate document. This document can be obtained by filing a written freedom of information request to Food and Drug Administration, Freedom of Information Staff, HFI-35, 5600 Fishers Lane, Rockville, Maryland 20857 and reference docket number 93D-0236.
Opportunities were also provided for public discussion and debate and these issues are also briefly summarized in this Executive Summary. To guide sponsors and enhance understanding of the value of individualizing therapy, this Executive Summary concludes with FDA recommendations for assessing gender effects.
David A. Kessler, M.D., Commissioner of Food and Drugs, opened the workshop. He noted that while the proportion of women participating in clinical drug studies generally reflected the prevalence of the condition under study in the general population, the safety and efficacy data from those clinical trials were not analyzed routinely for the specific influence of gender. He encouraged workshop participants to clarify critical and methodological questions aimed at improving and expediting the drug development process and to draw on FDA's 1993 "Guideline for the Study and Evaluation of Gender Differences in the Clinical Evaluation of Drugs" as a means of producing the safest and most effective medical therapies possible.
Janet Woodcock, M.D., Director of FDA's Center for Drug Evaluation and Research, then presented the formal workshop charge. She acknowledged that scientific knowledge and societal shifts have led to an understanding that the role of gender, coupled with other individual patient factors, should be evaluated during the drug development process. Dr. Woodcock noted that appropriate clinical trial enrollment in early drug studies, followed by rigorous data analysis, enhance substantially the ability to predict whether or not certain interactions are likely to be important to specific population groups. These predictions can then be examined in the overall database.
While the FDA's interest in assessing the effects of gender during drug development has been applauded in some quarters, it has also raised concern that too little is currently known about drug effects in women and the importance of those differences. Dr. Woodcock said that a substantial body of data about gender effects already exists, pointing out that in her own field of rheumatology, where a range of medical conditions predominantly affect women, most of the participants in clinical drug trials are women. However, she acknowledged that unanswered questions about how to assess gender effects in clinical trials remain.
Drug development sponsors and other researchers have raised concern about the practical and methodological consequences of assessing drug effects by gender. It has been suggested, for example, that the cost of clinical drug development could double if it were necessary to conduct parallel trials among men and women, each powered separately and each enrolling half as quickly as a less restricted trial. According to Dr. Woodcock, that concern has been fueled by the erroneous idea that information about drug effects derives solely from randomized trials. She pointed out that if every question about drug effects had to be answered via controlled trials designed specifically to answer that question, medical progress would slow dramatically. Rather, the goals of drug development science - to understand sources of variable response and causes of adverse reactions in order to allow more effective and safer treatments are supported by various kinds of data, including data on pharmacokinetics, metabolism, and drug and hormonal interactions, among others. A further approach is to pool data from multiple studies for demographic subgroup analyses. Appropriate clinical trial enrollment in early drug studies and rigorous data analysis greatly enhance the ability to predict when given interactions are likely to be important to specific population groups and, just as crucial, when they are not likely.
Dr. Woodcock cautioned conference participants that the deliberations of the workshop should not result in a rigid set of instructions for uncovering gender-related effects during drug development. Because the science is not yet mature, important questions remain about how best to detect those differences. She noted, for example, that under certain circumstances, early clinical pharmacology studies may be appropriately employed to elucidate gender differences, while in other instances large, simple trials that enroll many subjects with disparate characteristics may be more appropriate. The challenge is to determine the factors that may differentiate these cases to guide appropriate study design.
In outlining the structure and goals of the conference, Dr. Woodcock said that workshop findings are intended to assist both the FDA and drug developers to apply state-of-the- art scientific knowledge to the investigation of gender-related differences. She expressed the hope that further research and additional initiatives would result from the workshop and suggested that additional FDA recommendations may be forthcoming to guide sponsors on how to study gender effects during medical product development.
Women of Childbearing Potential and Early Clinical Trials
Ruth Merkatz, Ph.D., Director of FDA's Office of Women's Health, brought an historical perspective to the Gender Workshop, including a review of the FDA's 1977 guideline, "General Considerations for the Clinical Evaluation of Drugs," and the context in which it was issued. This guideline recommended excluding women with childbearing potential from participating in phase 1 and early phase 2 clinical studies until reproductive toxicity (segment 2) studies were conducted and some evidence of effectiveness had become available. The recommended exclusion was broadly applied to any "premenopausal female capable of becoming pregnant," but explicitly did not apply to women with life threatening diseases.
The policy was developed to reflect societal interests in protecting vulnerable populations and emerged in part from discoveries of birth defects resulting from fetal exposure to certain drugs, including thalidomide and DES. It also reflected the recognition that early clinical trials often take place prior to completion of preclinical reproductive toxicology studies, which include potential teratogenic effects on a developing fetus. With a possibility for fetal damage from experimental drugs and no clearly articulated medical benefit to the phase I and early phase 2 trials, women with childbearing potential were considered inappropriate subjects in early clinical trials. These considerations formed the basis of the exclusionary approach recommended in the 1977 guideline.
In later years, however, that recommendation began to raise important ethical and even legal questions about the appropriateness of assuming that women cannot take steps to avoid becoming pregnant, where appropriate, and of deciding for women that protecting the fetus outweighed other possible interests. Dr. Merkatz explored these questions in her talk and described the influence of the National Commission for the Protection for Human Subjects of Biomedical and Behavioral Research, which issued the Belmont Report in 1977, outlining the ethical principles expected to govern drug research. Subsequent interpretations of the report's emphasis on respect for persons, which encompasses the principle of autonomy, raised serious questions about the virtual ban participating in early clinical trials imposed on women with childbearing potential.
Further support for reconsidering the policy of excluding women with childbearing potential from clinical trials came from a number of women's health advocacy groups. These advocates argued that the policy had a chilling effect on access to later clinical trials, despite its focus on early phase trials. Despite the explicit exception for women with life-threatening diseases, there were also concerns that the 1977 guideline had an effect on this population as well. Because of accelerated approval and rapid development of experimental therapies to treat life-threatening diseases, it is important to include women earlier in these studies.
Policy Changes: The 1993 FDA Gender Guideline and NIH Initiatives
In 1993, FDA explicitly reversed the 1977 recommendation that women with childbearing potential be excluded from early clinical studies when it published "Guideline for the Study and Evaluation of Gender Differences in the Clinical Evaluation of Drugs." The new guideline also called for data to be analyzed to assess gender effect. Issues surrounding implementation of this guideline represented an important element of workshop deliberations.
The new guideline is best understood as part of a continuing effort to enhance individualized pharmaceutical therapy. This is based on the growing recognition that pharmaceuticals may need to be administered differently in different population groups identified by one or more demographic or clinical characteristics, such as gender, ethnicity, age, body size, hepatic or renal function, disease state or enzyme activity. Several important FDA documents reflect the early recognition of the value of individualized therapy:
Robert Temple, M.D., Associate Director for Medical Policy in the Center for Drug Evaluation and Research at FDA, explained how the new guideline was structured to promote individualization of drug therapy. Like the guideline focused on the elderly, the gender guideline states FDA's expectation that the data supporting an NDA will include a reasonable representation of the population that will receive the drug, and that safety and effectiveness data will be examined to identify significant differences by demographic groups, such as gender and age. Dr. Temple explained that the 1993 Guideline focuses on data needed for approval and marketing, with only limited reference to the details of development or when and how a sponsor should study both genders. He emphasized that rigid gender quotas are not expected in clinical trials but that the guideline does express a preference for including both genders in the same studies, rather than in separate single-gender studies. It also suggests that FDA might require the inclusion of both genders in early safety, efficacy, and dose-finding studies where drugs for serious or life-threatening diseases are likely to be made widely available prior to approval, or are Rely to be approved on the basis on phase 2 data.
The 1993 Guideline emphasizes the process of informed consent so that trial participants understand what is known and unknown about experimental agents. It stresses the need, where appropriate, for women with childbearing potential to use contraception or abstinence while participating in early trials. It recognizes as well that under certain circumstances pregnancy tests may be an important part of protocol design in order to guard against the potential for fetal damage. The 1993 Guideline thus reflects the FDA's continued interest in protecting the fetus from unanticipated exposure to potentially harmful drugs, while bringing renewed attention to the benefits that may accrue to women when they are able to participate in all phases of drug development.
More specifically, the new Guideline describes the importance of pharmacokinetic studies, the possible contribution of factors other than gender to an apparent gender difference (such as body size, smoking, and concomitant illness), the value, and sometimes the difficulty, of conducting relevant pharmacodynamic studies, the value of population pharmacokinetic and pharmacodynamic analysis (a pharmacokinetic screen, in FDA parlance) and the importance of conducting relevant subset analyses. In addition, the Guideline indicates that the influence of menstrual status, that is, whether women are pre-or post-menopausal, and menstrual cycle on the pharmacokinetics of an investigational drug should be explored, as should the effects of concomitant supplementary estrogen treatment or systemic contraceptives. Finally, the Guidance suggests that the influence of the investigational drug itself on the pharmacokinetics of selected oral contraceptives should be explored.
A primary focus of the Guideline is to recommend that analyses be performed to assess differences in drug action due to gender in controlled clinical trials conducted to establish safety and efficacy in the total population. Such analyses may be presented as integrated summaries, which are essentially meta-analyses, in which data from multiple trials in a clinical trial base are combined and subjected to statistical analysis for gender effects. Because of their post-hoc character, these analyses do not test a hypothesis but instead are descriptive, a characteristic they share with most analyses of safety data. Clearly, such analyses can support further studies but if the information from these analyses is consistent, it can also be incorporated into product labeling and be useful to patients and practitioners even without further studies.
Although current FDA regulations do not require the participation of specific patient subsets in particular trials, a proposed new rule requires that the NDA format and content include presentations of safety and effectiveness data for important subgroups, specifically gender, age, and racial subgroups and, when appropriate, other demographic subgroups. These proposed new rules will also request that the IND annual report include information about the total number of subjects enrolled into clinical trials, categorized by gender, age, and race. [Docket 95N-00101]
Several workshop speakers emphasized that FDA's 1993 Guideline has had a clear impact on clinical drug trials. Janice Bush, M.D., Vice President of Janssen Research Foundation, said that since its publication, pharmaceutical sponsors have enrolled more women and included them earlier in the drug development process. Dr. Merkatz also emphasized the progress that had been made in articulating the guideline principles to FDA staff, as well as to sponsors of research, scientific and academic groups, health advocacy groups, and other constituencies. She noted that special attention has been focused on the Institutional Review Boards, which review protocols, and appropriate inclusion and exclusion criteria and informed consent procedures have been emphasized.
Significantly, as issues of access to clinical trials. were being debated at the FDA, initiatives at the National Institutes of Health (NIH) were moving in a similar direction. Eugene Hayunga, Ph.D., Research Policy Officer at the Office of Research on Women's Health (ORWH), described NIH policy to workshop participants. While NIH has required that women and minorities be included in clinical research since 1986, a series of policy revisions in the early 1990s strengthened this mandate. The NIH Revitalization Act of 1993 then gave these policies the force of law. A year later, a new guideline was issued jointly by ORWH and NIH's Office of Research on Minority Health.
The guideline requires the NIH to ensure that women and members of minority groups and their sub-populations are included in all NIH-supported human subjects research and in phase 3 clinical trials in numbers sufficient to allow a valid analysis of differences in intervention effect. Researchers are not allowed to use cost as an acceptable reason for excluding these groups. Also, the NIH must initiate programs and support for outreach efforts to recruit these groups into clinical trials.
Together, the 1993 FDA guideline and the NIH initiatives point the way for greater analysis of the gender-related effects of medical products. Nonetheless, the Guideline continues to generate comment and debate as scientific, ethical and liability exposure issues are considered. Dr. Bush commented that industry needed to make better use of the data on potential gender effects that is now being collected. She noted that the workshop could make a useful contribution by offering practical proposals that did not impede clinical research or impose data requirements without sound scientific justification. Also essential to the ongoing dialogue is a frank discussion of liability, said Dr. Bush. While there is little record of substantial liability claims by clinical trial participants, children adversely affected by the medical decisions of a parent have the right to pursue litigation when they reach the age of majority in many states and this is a cause for concern.
Some patient advocacy groups continue to claim that legal and medical obstacles to trial participation are still imposed on women with childbearing potential, despite the guideline changes. In her workshop presentation, Theresa McGovern, Executive Director and counsel to the HIV Law Project, suggested that the obstacles may be particularly great for HIV-positive women. Ms. McGovern offered specific examples of clinical trials from which this population had been excluded, as well as instances in which the side effects of approved drugs were poorly understood in HIV-positive women. That perspective was echoed by a patient spokesperson, Ms. Claudette Bain, who described her concerns about various AIDS therapies that had been minimally tested in women. Jeanne DeJoseph, Ph.D., another patient spokesperson, discussed the relative lack of safety and efficacy drug profiles for premenopausal women who had suffered a myocardial infarction.
With the historical and regulatory background clarified, the Gender Workshop's three panels - Pharmacokinetics and Pharmacodynamics; Hormonal Influences; and Study Design and Analysis - were convened. During each panel, speakers briefly summarized pertinent research, issues and conclusions. A moderator-led panel discussion followed each set of speaker presentations, allowing relevant subjects to be developed further. The discussions of the three panels and their recommendations for assessing gender differences during medical product development are summarized below.
Panel 1: Pharmacokinetics and Pharmacodynamics
Moderator: Alan Sedman, M.D., Ph.D.
Panel I speakers discussed the following issues: Review of Published Data Regarding
PK/PD by Dr. Janice Schwartz, Regulatory Aspects of Gender Based PK/PD Differences
by Dr. Mei-Ling Chen, Examples of Gender Based PK/PD Differences by Dr. Raymond
Woosley and A New Approach to Drug Development by Dr. Lewis Sheiner.
The pharmacokinetics and pharmacodynamics panel discussion focused on possible
clinical development strategies to assess gender effects, using nonclinical
data for two hypothetical drugs, termed A and B.
Drug A: Drug A is an inhibitor of plasma renin activity in development to treat hypertension. In hypertensive rats and dogs, this compound demonstrated gradually increasing activity over a broad range of doses. Toxicity was not evident in animals at doses several orders of magnitude greater than those expected to be therapeutic. Oral drug absorption was highly variable and absolute bioavailability averaged five to 10 percent in animals. Binding to human plasma proteins was negligible. Renal elimination of unchanged drug predominated but approximately 30 percent of absorbed drug was eliminated in urine as metabolites. Human liver preparations suggested that CYP2D6 is a potentially important isozyme in the metabolic elimination of the drug. Gender differences in animal toxicokinetic studies were not observed.
Drug B: Drug B is a selective Ml agonist being developed to prevent or provide symptomatic treatment of Alzheimer's disease. In preclinical tests in rats and monkeys considered predictive of cognition in humans, the compound markedly enhanced performance over a narrow range of doses. Intractable seizures were evident in animals at doses six to eight times above those expected to be therapeutic. Oral drug absorption was very reproducible and absolute bioavailability averaged 80 percent in animals. The drug was extensively bound to human plasma alpha,-acid glycoprotein (97%) and was metabolized to numerous moieties, with less than 0.5% of the dose recovered unchanged in urine. Human liver preparations suggested that metabolism occurs via CYP1A2 and CYP3A4 isozymes. In rats, marked gender differences were observed in toxicokinetic studies. The plasma concentrations of unchanged drug in females were six times greater than those in males. Approximately two-fold differences in the same directions were observed in monkeys.
Panelists discussed the preclinical studies for both Drug A and Drug B, noting that Drug A studies suggested little potential for gender-related clinical differences while the preclinical data for Drug B suggested that responses in men and women were likely to differ. Both cases demonstrate the value of preclinical studies to assist the efficient design of further clinical investigations In vitro plasma protein binding and hepatic tissue metabolic profiles can be utilized to determine the likelihood of significant gender-related differences in human Pharmacokinetics. Pharmacokinetics, pharmacodynamics, pharmacology, and toxicology information in both male and female animals can also suggest potential gender-related differences in concentration response, safety, and/or efficacy.
The panel recommended use of animal models with metabolic pathways similar to humans when possible. Attempts should be made to develop preclinical models to understand the gender-related pharmacokinetic and pharmacodynamic differences observed in humans. Acceptable preclinical models that assess the effects of hormones on drug metabolism, pharmacokinetics, or Pharmacodynamics are still lacking. Preclinical and in vitro information regarding drug metabolism, such as the cytochrome P450 isozymes responsible for biotransformation, should be especially useful in determining whether drug-drug interaction studies between a new drug and hormone replacement therapy or oral contraceptives are Rely to demonstrate significant potential for metabolic inhibition. For drugs to be used by both men and women, the panel recommended that both genders be included in early clinical trials to screen for potential differences in pharmacokinetics, pharmacodynamics, tolerance and/or efficacy. If preclinical data suggest little potential for gender effects and no differences are observed in initial human trials, additional trials specifically to explore gender effects may not be indicated. If preclinical or early clinical information suggests a high likelihood of gender differences, a further series of carefully designed studies should be considered to determine whether the drug needs to be used differently in men and women. Such studies would usually be performed prior to large pivotal efficacy studies and would carefully compare the pharmacokinetics, Pharmacodynamics, safety, and/or efficacy of the compound in men and women, possibly distinguishing between pre- and postmenopausal women. For almost all drugs, the panel believed it would be useful for phase 2 and 3 clinical pharmacology databases to be analyzed using population approaches to look for potential gender-related differences of clinical significance, as well as for age, race, and disease state, on response, unless discrete studies of these factors have been already conducted and analyzed.
In summary, the panel concluded that the potential for clinically significant, gender- related differences in pharmacokinetics, Pharmacodynamics, safety, and/or efficacy should be considered early in drug development. Preclinical and early clinical information is critical for optimal design of early and late phase clinical trials and to determine whether special studies in humans are needed to define gender-related differences in clinical response. All studies in all phases of drug development should be carefully designed to answer specific sets of questions, rather than based on standard protocols and test methods.
Panel I Recommendations
1. Acceptable preclinical methods to assess the effects of gender and gender-related factors, such as hormonal influences on drug metabolism, pharmacokinetic, or pharmacodynamics, are still lacking and should be developed.
2. Clinical pharmacology databases should be examined to identify potential gender-related PK/PD differences of clinical significance.
3. Where appropriate, phase 2 and 3 databases available to FDA should be analyzed to look for gender effects, as well as the effects of age, race and disease state, on safety and efficacy, unless discrete studies of these factors have already been conducted and conclusions drawn.
4. Pre-clinical and early pharmacokinetic and/or pharmacodynamic information should be provided to allow optimal design of subsequent clinical trials and to determine whether special studies are needed to define gender-related differences in clinical response.
5. Studies in all phases of drug development should be carefully designed to answer specific sets of questions based on prior knowledge of the intended use and therapeutic range of the drug under investigation.
Panel Discussion II: Hormonal Influences
Moderator: Jean P. Rowan, M.D.
Panel II speakers discussed the following issues: General Physiology of Female Hormones over the Lifespan by Dr. Edward Wallach, Important Hormonal Influences on Drug Metabolism by Dr. David Flockhart, Hormonal Influences on Metabolism of Cardiovascular Drugs by Dr. Paresh Dandona, Hormonal Influences on Metabolism of Psychotropic Drugs by Dr. Jean Hamilton and Methodologies for Studying Hormonal Influences by Dr. Scott Lukas.
There is substantial data to show that endogenous and erogenous steroid hormones affect the kinetics and dynamics of drugs. The panel on hormonal influences structured its discussion around four questions as it tried to determine the clinical relevance of these PK/PD changes:
1. What mechanism of drug action should prompt an investigation of hormonal influences?
2. What enzyme systems are apt to be influenced by changes in hormonal environment, and thus impact the safety and efficacy of a drug?
3. In clinical trials, what findings suggest hormonal influences may be active?
4. Should studies on endogenous and erogenous hormonal influences be conducted as phase I studies, or confined to studies of women under treatment in phase II and III trials?
The panel concluded that pharmacodynamics, rather than pharmacokinetics, more accurately predicts clinically important safety and efficacy effects. It also agreed that the mechanism of action of the gonadal steroids is not well understood, and that side effects from drug therapies may often be key indicators of an underlying hormonal influence. The panel noted that therapeutic agents can have a variety of unanticipated effects on estrogen-responsive tissues. Often, such tissues are not traditionally considered "target tissues" of either the gonadal steroid hormones or the agents themselves; blood vessels are an example. The panel also discussed the effect of the various isoenzymes in the CYP 450 system - for example, the 1A and 3A systems. While there are relatively small changes (approximately 20 to 30 percent) for the cytochromes under discussion, these cytochromes have a broad range of metabolic activity, e.g., the 1A and 3A system, at the extreme end, the difference between women and men is relatively large and might well be clinically relevant. This is an important area for development of preclinical screens of drugs using the CYP 450s of steroidogenic enzymes, similar to those used for hepatic CYP 450 isozymes, to study how drugs can interfere with the endocrine system.
Side Effects: The panel noted that certain drug side effects may indicate an underlying hormonal interaction. A variety of mechanisms need to be examined carefully, and compartmentalized by possible site of action to unravel the relationships that occur and to determine the differing effects that may be produced by the same drug. Drugs that have a small margin of safety, or serious adverse effects, warrant special scrutiny.
The tendency to ignore what patients say, particularly in terms of side effects, was noted as troubling. As the data presented at the workshop demonstrate, women have vascular systems that are, in many ways, different from men and that may result in such side effects as light-headedness, headaches, and nausea. Another side effect, and one that is often overlooked when drug effect is considered, is change in the menstrual cycle, such as amenorrhea or excessive bleeding. Mood swings and depression may also suggest a hormonal response that merits a closer look.
The data also suggest that women are more likely to have side effects if the vasodilatory effect associated with the drug is very great. In that context, it is important to ask whether there are any data to show that the flushing, dizziness, throbbing headaches, and ankle swelling associated with certain drugs, such as calcium-channel blockers, are more common in women. This may prove relevant to a finding several years ago that grapefruit, which contains a flavonoid called naringin, was shown to inhibit the metabolism of nifedipine, increasing the drug's bioavailability. This interaction may produce more marked side effects in women. The possible role of estrogen in this reaction needs to be examined.
Studies with the dihydropyridine calcium channel blocker, amlopidine, found three drug-related side effects - flushing, edema, and palpitations - were considerably more common to women than to men. Used at a dose of 15 milligrams, amlopidine gives a 30 percent rate of edema, which is high By contrast, a 10 milligram dose has a low edema rate. Possibly, smaller body size puts women a little further out on the dose response curve, but other basic physiological or metabolic factors may also be involved.
Unanticipated Drug Effects on Estrogen-Responsive Tissues: A variety of mechanisms need to be examined carefully and compartmentalized by the site of action. Many agents exist with unexpected side effects on the reproductive system. One important realm of scrutiny is the central nervous system, and drugs acting at the level of the central nervous system to influence hypothalamic function. Signals governing hypothalamic synthesis, storage, and secretion of GnRH involve adrenergic, serotonergic, and dopaminergic mechanisms that influence GnRH output and, ultimately, ovulation. One possible example are the psychopharmacologic agents that give rise to hyperprolactinemia at this level, and ultimately suppress ovulation, causing amenorrhea and bone loss.
Over-the-counter anti-inflammatory agents provide another example of unexpected
side effects on the reproductive system. The mechanism of ovulation has been
Uened to an inflammatory process by a number of researchers, with release of
histamine, prostaglandins, especially F2 alpha, and oxygen-free radicals. Nitric
oxide has also been implicated. Cyclo-oxygenase inhibitors and anti-prostaglandins
are commonly used over-the- counter agents that may, in certain dosages, influence
ovulation by acting locally at the follicular level.
Another unexpected cross-reactivity between drugs and hormones may occur when
drugs and hormones share, and compete for, the same receptors. For example,
spironolactone taken by pregnant women competes for, and blocks, androgen receptors,
profoundly influencing intrauterine development of male external genitalia.
In a related
example, tamoxifen blocks the estrogen receptor when given to treat breast cancer
but also acts on estrogen receptors in the endometrium to increase the risk
of endometrial cancer. The manufacturer claims tamoxifen also supports non-destruction
of the bone.
The mode by which estrogen acts at the bones is not precisely understood. Mononuclear cells from post-menopausal women generate more cytokines, such as TNF-alpha, IL-1 and IL-6, than from pre-menopausal women. When post-menopausal women are treated with estrogens, their cytokine levels return to normal. This is interesting because the circulating monocyte macrophage is a cousin of the osteoclast, which is a phagocytic cell that breaks down bone. Moreover, the so-called osteoclast activating factor has been shown to be TNF-alpha.
The steroidogenic CYP 450 enzymes are certainly affected by drugs, including ketoconazole, which inhibits steroidogenesis. Recent clinical evidence also points to griseofulvin as affecting steroid metabolism. Women treated with griseofulvin for toenail fungal disorders develop dysfunctional anovulatory bleeding, which stops when griseofulvin is discontinued. Post-menopausal women on hormone replacement therapy also experience this side effect.
Finally, drugs that affect vascularization or neurovascularization may interfere with corpus luteum function, since the function of the corpus luteum is to develop a transient neurovascular structure from which progesterone is transported.
Clinical Endpoints in Pharmacodynamics: The discussion regarding the nature of clinical trial findings, which might suggest that hormonal influences are active, reflected a broad understanding of the role, and mechanism of action, of the gonadal steroids. The knowledge that erogenous estrogens in the postmenopausal woman affect many different systems is apparent in the treatment of vasomotor symptoms, vaginal atrophy and dryness, and osteoporosis. Strong epidemiological and clinical information, based on surrogate endpoints, demonstrates that estrogens are active in cardiovascular health and in preventing Alzheimer's-type dementia. If exogenous estrogens in the postmenopausal period are active in all these systems, then endogenous estrogens almost certainly are active in those systems prior to menopause. As such, endpoints become dynamic indicators of hormonal influences. Certainly, when studying drug mechanism of action, and drug activity in disease areas such as those just mentioned, both endogenous and erogenous sources of hormone must be considered.
The menstrual cycle is such a sensitive mechanism during reproductive life that menstrual irregularity may be an early indicator of a drug effect. Often, information about a subject's menstrual cycle is sparse or unavailable but it is a potentially useful data source that should perhaps be collected more rigorously. In a woman, the clinical evidence to examine are menstrual irregularities, amenorrhea, failure to conceive despite attempts to do so, and conception and early pregnancy loss, especially if that loss is associated with a structural or chromosomal abnormality. Because males have no comparable indicator, it may take longer to know whether a drug has a damaging impact on the hypothalamus and pituitary.
One panelist emphasized the need to look for the source when results differ between men and women. Likewise, any time a statistician finds considerable variability, gender effects and underlying hormonal mechanisms should be considered. Although pharmacokinetic differences, per se, are not a reason to pursue gender studies, a disassociation between the pharmacodynamics and the pharmacokinetics suggests there may be an underlying hormonal difference, or an interaction of endogenous or exogenous hormones, of interest. Any changes in drug efficacy, or anything with a marked effect on dose ranging or the side effect profile, should immediately lead to an exploration of hormonal influences.
As clinicians, there is a great need to look at new ways to optimize treatment. In an emerging area such as gender effects, it is important to keep an open mind, and pursue investigation, even when some of the effects initially seem modest. Dosing changes of anti-depressants during menstrual cycle illustrate the need to define clinically sensitive sub-populations, and optimize therapies for them. It is also important to look at variations within each gender group and across genders. In many dose response trials, it's not easy to identify patterns, either in measures of effectiveness, or in side effects. They are probably most significant when the dose response curve is reasonably steep within the therapeutic range. Where titrated doses are used, each patient becomes his or her own dose-response trial and the requirements for eliciting gender differences become very different.
Timing of Gender Studies: The panelists discussed whether studies of endogenous and exogenous hormonal influences should be conducted as phase I studies, or confined to phase 2 and 3 trials. They agreed that case-by-ease decisions are needed and must be driven by the science. As the knowledge base expands, and the in vitro and in vivo areas in which generalization is, and is not possible becomes clearer, studies can be expected to be more focused. While more definitive generalizations may be possible in five years, a few guidelines were proposed by the panel:
1. If there is primary metabolism through CYP 450 la or 3a, kinetic effects are more likely, and gender and hormonal influences should perhaps be considered in phase I. Other factors also come into play, including high first pass metabolism through the liver, and great variability in bioavailability.
2. If there is a very strong suspicion that hormonal effects exist, from metabolic and enzymatic standpoints, as well as from prior experience in that class of drugs, it may be highly desirable to establish those effects early in drug development.
3. Because the diverse side effects that indicate hormonal influence are so much greater in women than in men, they can be very difficult to predict and use in phase I testing. From that perspective, phase 2 and 3 may be more appropriate places for testing hormonal influences.
4. There is a need to study drug interactions, particularly for drugs that have predictable effects, such as the oral contraceptives, hormone replacement therapy, danazol for endometriosis, and estrogen therapy for prostate cancer.
5. Menstrual cycle data should be collected routinely from female subjects.
Panel II Recommendations
1. Metabolic pathways should be described during the preclinical phase of development.
2. If metabolism of an investigational agent occurs primarily through CYP 450 IA2 or 3 A4, pharmacokinetic differences due to hormonal influences are more likely; this influence should be examined in early drug testing.
3. Although blood vessels have not traditionally been considered target organs for estrogen action, a number of studies support the finding of intrinsic vascular regulatory mechanisms that are responsive to estrogens. Further research in this area should be conducted.
4. The mechanism of action of the gonadal steroids is not well understood. Side effects from drug therapies may often indicate an underlying hormonal influence. Drugs can have a variety of unanticipated effects on estrogen responsive tissues. Therefore, pharmacodynamic differences between genders, rather than pharmacokinetic differences alone, should be studied because the former predict clinically important safety and efficacy effects more accurately.
Panel Discussion III: Study Design and Analysis
Moderator: Murray Lumpkin, MD.
Panel III speakers discussed the following issues: Subsets: Opportunities and
Cautions by Dr. Robert Temple, Statistical Aspects of Assessing Gender-Related
Treatment Specificity by Dr. Richard Simon, Survey of Gender Analyses in the
NDAs for Selected New Molecular Entities in 1993 by Dr. Stella Machado, Methodological
Considerations for the Safety and Evaluation of Gender Differences in the Clinical
Evaluation of Drugs by Dr. Robert O'Neill, Prospective Studies: Issues and Rationale
by Dr. Roger Williams, Pharmacokinetic Screen: What Can Be Accomplished by Dr.
Jean Louis Steimer, FDA Gender Guideline: Perspective of an Industry Statistician
by Sarah JH Kogut and
Special Consideration in Using Cardiovascular Devices in Women by Dr. Julie
Swain.
In an effort to learn more about study design and analysis, this panel focused on four key questions:
1. What types of results in early phase studies imply the need for more intensive evaluation of potential gender differences in phase 3?
2. When and how should trials be designed to determine if different dose regimens are needed in women?
3. When are separate, fully powered trials needed to gain safety and efficacy information about gender differences?
4. Does it make a difference in planning, if a potential difference in safety, rather than in effectiveness is possible?
Panelists agreed that early indications of gender differences in PK studies are signals that gender effects should be considered in later trials. Any PK non-linearity difference is a cause for concern although it is rare that investigators will study different regimens in men and women to determine this possibility. In general, clinicians assume there will be no efficacy differences by gender unless available data shows an important interaction.
There was broad agreement that very few studies are designed to optimize the dose in individuals, that most studies are analyzed by group data, and that investigators need to pay greater attention to dose-response at all phases. Given the complexity of the related issues, the value of bringing together people from different disciplines and with differing perspectives was noted by several panelists. The dose-response issue becomes most important for drugs with narrow therapeutic indices, especially those accompanied by steep dose-response curves. Such situations generate a lot of overlap in the data and are characterized by many confounding variables that make it hard to determine dose- response differences. Very often, PK and PD screens can help sort out these differences. Such analyses may be performed using sparse data from late phase studies to yield valuable information about gender and other effects that would be otherwise difficult to determine.
The relative merits of determining dose response in phase 2 and phase 3 trials were discussed extensively. An industry spokesperson said that one reason for determining dose-response only late during phase 3 trials was to respond to the pressure both from the public and from private industry to develop drugs as quickly as possible. From industry's perspective, it is important to conduct studies in a certain sequence in order to acquire the information necessary to move onto the next step. If dose estimates are not established until later in drug development, this approach saves time, although it also involves risk. If there is some reason to expect toxicity, and the dosage is not going to be titrated, perhaps because the adverse effects are sudden, securing good dose findings in phase 3 trials becomes especially important. Another speaker noted that while every available pharmacologic index or indicator of potential effectiveness should be used in phase 2, these studies are unlikely to identify the correct dose because the endpoints of interest are not used and the trials usually aren't sufficiently large. For example, the dose of drugs designed to increase cardiac output or reduce symptoms of heart failure are not necessarily the doses that produce the best survival outcome. In addition, safety findings during phase 2 are too infrequent to yield much information about gender-specific differences.
Nonetheless, there are many limitations to doing dose-response studies in phase 3 and some arguments for gathering the information earlier. Some panel members noted that it is more cost-efficient to determine dose-response relationships in phase 1 and phase 2 and that this allows for a fuller understanding of safety and efficacy profiles prior to phase 3. Panelists generally agreed that as much dose-response information should be collected as early as possible. Certainly, the pharmacokinetic screen should not be confined to late phase trials, especially because PK data obtained earlier can provide significant information relevant to decisions about dose-response. On a related topic, panel members urged sponsors to encourage more communication among the clinicians, scientists and statisticians involved in both phase 2 and phase 3 studies. The necessity of enhanced communication was highlighted by two recent trials that resulted in unexpected findings at the end of phase 3. This may have been avoided by a more careful review of phase 2 studies, where the data clearly showed significant blood level differences between study groups.
In reality, most studies are not classical dose-response studies but can best be described as "find me a winner among one of the doses." For example, in studies of nucleoside analogues, the low dose was preferred because of toxicity at high doses. In terms of individualized dosing, dose-response studies might be designed to use two different high doses and two low doses, for males and females. This provides a variation on dose-response for males and females, since one need not look for a smooth curve but rather a curve that maximizes the efficacy/toxicity ratio.
Despite the challenge of determining individual dose response relationships, several concrete strategies were proposed. Dr. Sheiner has written about an approach to dose finding in which individuals are titrated to a therapeutic endpoint. This technique can be applied to conditions that are relatively responsive and reversible. Careful analysis, using NONMEN, is required when trying to relate dose to response, because those receiving higher doses tend to fare less well. This approach offers an opportunity to develop an initial idea of the dose range and may offer advantages over trials in which a single dose is given to all subjects. A specific goal, therefore, is to develop study designs that generate interpretable ways of titrating dosages.
Another proposal for determining the proper dose level is to develop a table showing the percentage of patients who gain the intended benefit, with an acceptable toxicity level, at each dose level or range of dose levels. Such a table can be used for all patients, hopefully including a representative sampling of women and men and some ethnic diversity. If dose levels prove not to matter much, and a fairly high number of patients benefit from the drug, then a single fixed dose level can be chosen. If the dose ranges vary substantially but patients benefit from the drug, the information can be used to establish an appropriate dose level.
Panelists also suggested the savings generated by greater flexibility with dosage should be considered. Greater flexibility in dosing, as defined during studies of gender-based dose differences, may provide savings of benefit to practitioners and consumers.
Another possible benefit is the opportunity to use advertising to highlight dosing that reflects gender-specific differences in efficacy. The need for early recognition of gender differences in device use was also discussed. In general, little attention has been paid to these differences by either the device industry or the FDA. Panelists called for the FDA to take the lead in promoting awareness of this issue, perhaps by issuing guidelines.
Panel III Recommendations
1. More complete by-gender tabulations and graphics of safety and efficacy results are needed, as demonstrated by the results of an internal FDA study showing that three of 15 new molecular entities approved in 1993 did not include a gender analysis. In addition, more analyses of combined data sets are needed and new ways should be found to facilitate meta-analyses when combining studies with varying designs.
2. Although, suitable study designs allow subset differences in safety and effectiveness to be identified and rational dose labeling provided, strategies for improving the methodology are needed. Early indications of gender differences in PK studies should signal the need to examine gender effects more closely in subsequent trials and offer the prospect of improving late phase clinical trial designs, possibly saving time and money.
3. Good population and individual dose response curve information for safety and efficacy effects is critical to establish the need for different dose regimens between genders. Gender-specific dose response information can be determined in phase 2 and phase 3 studies.
4. Gender analyses in large-scale clinical drug trials have often been performed as tests of "interaction" in the analysis of variance. Such tests have low power, compared to tests of the main treatment effect, i.e., the study would have to be at least four times its planned size to achieve the same power. More information is obtained when separate statistical tests are performed on mates and females, but even these tests are less powerful than the test for the effect in the whole population. Thus, it will not be unusual to fail to see a significant effect in either subgroup. The panel recommended that confidence intervals for the treatment effect should be calculated for males and females separately, and compared with the confidence interval for the treatment effect for the whole population. Graphs of the confidence integrals are helpful to show consistency between genders and the amount of certainty in the findings are readily apparent. Bayesian approaches may also be useful in this context.
5. When combined with phase 3 and other data, population PK/PD methods may be useful to enhance understanding of gender effects, and in particular, how gender, age, race, and other patient factors may be interrelated.
6. Generally, designing a larger clinical trial or combining databases can provide more definitive subset information from trials. Although the quality of inferences drawn from a single study may be better than from a combined data base of equivalent size, there are a variety of reasons that enlarging the size of a single study to increase the power of separate analyses may be less attractive. Relevant factors include the drug and the condition being treated and certain ethical constraints may also come into play. For example, the possibility of an early halt to a mortality study may preclude collecting sufficient data on both genders. On the other hand, meta-analyses should be conducted with care, especially when the studies to be combined have different designs. Graphical methods are a helpful adjunct to meta-analyses.
7. Where information about potential safety and efficacy gender effects of drugs has been identified, it should be better communicated among sponsors, FDA review- ers, health care providers, and patients.
8. Earlier recognition of gender differences during the evaluation of medical devices is needed. FDA should take the lead to help promote awareness of this problem.
In an effort to hear a broad range of opinions, FDA accepted public comment on the workshop topics in an open hearing facilitated by Dr. Ruth Merkatz, Dr. Roger Williams and Dr. Elaine Esber. The four speakers who presented testimony each emphasized the importance of understanding gender-related differences.
Dr. Elizabeth Lane of the Pharmacokinetics Laboratories reported on a Medline search in which she found that a higher clearance or lower bioavailability in women was reported for II drugs and with a ratio of greater than 1.5 in eight drugs. However, she suggested that the higher clearance appeared to be an artifact of smaller body weight in women.
Dr. Anne Nafziger of Bassett Healthcare observed that sponsors continue to be reluctant to include women with childbearing potential in all phases of trials yet exclusion from early trials impacts on dose finding and epidemiological studies of drug toxicity in women. Even the studies that generate relevant data on gender-related drug effects are rarely designed with that aim and as a result are underpowered to identify important gender differences (e.g., whether menstrual cycle has a clinical impact on PK/PD). On another topic, Dr. Nafziger urged authors to facilitate literature searches dealing with gender differences by indexing their publications using a standard key word recognized by Index Medicus.
Dr. Naomi Kaminski of the American Pharmaceutical Association noted that clinical literature tends to combine data from men and women despite the gender-related differences that have been observed during research. Pharmacokinetic and pharmacodynamic studies should result in relevant data so that the pharmacist understands a medication's effect on women and the impact of oral contraceptives and hormone replacement therapy. The speaker described a project of the American Pharmaceutical Association in which a research network of pharmacists is being established to participate in postmarketing studies that examine gender differences.
Ms. Julie Davids of ACT UP, Philadelphia urged that a regulation be issued quickly requiring women to be included in trials for serious or life threatening diseases. (On September 24, 1997, a proposed regulation to address this issue was published in the Federal Register.) She also asked attendees to consider research with people who are taking female hormones, noting that transsexuals who use these hormones are underrepresented in epidemiological research.
FDA RECOMMENDATIONS: STRATEGIES FOR ASSESSING GENDER EFFECTS
Based on the deliberations of the 1995 Gender Workshop, two central questions for assessing gender effects during drug development emerge: 1) How can we best deter- mine whether the dose regimen should be adjusted based on gender? 2) When during drug development, should this determination be made? The answers to these questions depend on the specific drug and its intended use, the degree of certainty that must be achieved, and the information available about the drug itself or related drugs. The FDA is involved in probing these questions further in order to develop strategies that will assist sponsors and other researchers to analyze gender-related drug effects which provided the impetus for this workshop.
As noted by the Workshop participants and delineated in the 1993 Guideline, information about potentially important gender effects can be provided by a number of clinical and non-clinical approaches, including:
in vitro studies in human or non-human tissue;
in vivo studies in non-human species;
early phase exploratory clinical studies using pharmacokinetic and pharmacodynamic approaches, coupled with a mechanistic understanding of drug action, if available;
late phase confirmatory studies in humans;
post-marketing studies.
A combination of these approaches is likely to prove useful in defining gender and other population subset effects. To build on the data already being collected in clinical trials, the need is also apparent for: 1) more complete tabulations and graphics of safety and efficacy data grouped by gender; 2) more by-gender analyses, generally by greater use of meta-analysis to maximize the amount of information gained for both genders; and 3) new ways to think about and perform meta-analyses of information from studies of varying design.
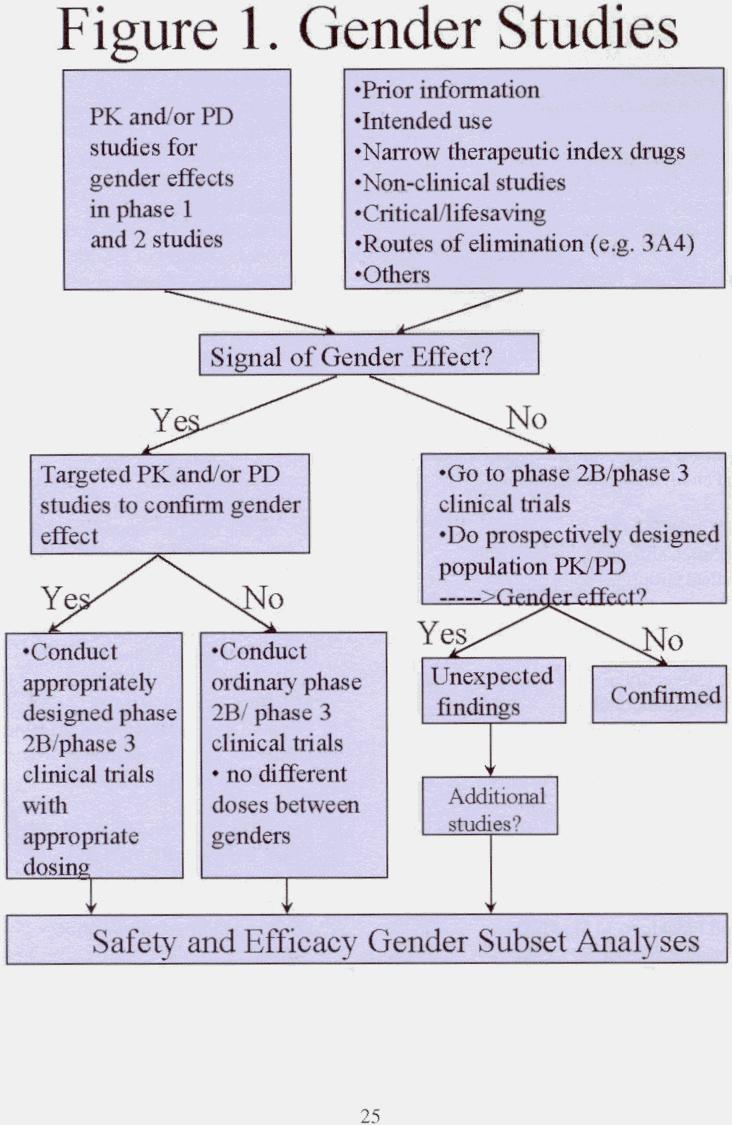
A 'decision tree' approach to thinking about ways to assess gender effects during the development of medical products may be useful (Figure 1). At the start of clinical development, prior information - such as the intended use of the drug, the likelihood of the drug having a narrow therapeutic range, the drug's possible routes of elimination, its use for critical and lifesaving illnesses, animal PK/PD data, and other pertinent factors - should be coupled with initial PK/PD studies to determine whether signals of a gender effect exist. Included in this initial screen might be data from non-clinical studies, such as in vitro studies of human tissue, which are routinely used to identify possible important routes of metabolism and potential drug-drug interactions and which might signal the presence or absence of a gender effect. Workshop discussions suggest, however, that further work may be necessary before this approach can be used to predict gender effects reliably. Some studies do suggest, however, that if significant metabolism occurs via CYP 450 1A and 3A, a gender and/or hormonal effect may be more likely.
In early phase clinical trials, entry of sufficient numbers of both genders will allow initial pharmacokinetics and perhaps pharmacodynamic evaluation for a gender effect. In general, important pharmacokinetic differences by gender is readily obtained even when the numbers of subjects in these studies is relatively small. Where good pharmacodynamic measures exist (as they do, for example, with beta-blockers, ACE inhibitors and others), it is relatively easy to obtain a good pharmacokinetic/pharmacodynamic relationship that allows important clinical differences to be anticipated. The availability of a suitable pharmacologic endpoint to allow a pharmacodynamic or other clinical assessment of a gender difference may be problematic, although recent surveys per- formed by FDA suggest that some useful pharmacodynamic endpoints may be available in up to 50 percent of investigational drugs, and possibly more. If a signal of gender effect emerges, more elaborate pharmacokinetics and pharmacodynamic studies may be indicated. An early indication of by-gender differences, subsequently confirmed by targeted pharmacokinetics and/or pharmacodynamic studies, suggest that later phase studies should be designed and powered to assess gender effects. If pharmacokinetics, and, if possible, pharmacodynamic results from early studies are negative, subsequent reliance on population pharmacokinetic/pharmacodynamic approaches (pharmacokinetics screens) and post-hoc gender subgroup analyses should generally be sufficient to investigate the impact of gender on the safety and efficacy of a drug.
The post-hoc subset analyses now relied on by the FDA to assess gender and other subset group effects are likely to detect large differences in safety and efficacy, but smaller differences may go undetected. A remaining question, then, is whether smaller differences - which might be discovered from more precise pharmacokinetic and pharmacodynamic studies, should be translated into labeling instructions. The answer will be made on a case-by-case basis, recognizing that the population dose response relationship and the steepness of the dose/response curve will determine whether different dosing recommendations should be provided to men and women.
The 1995 Gender Workshop provided an opportunity to review the 1993 Guideline and to consider the need to provide further clarification. While there is now a widespread consensus that important scientific questions about gender-based biology and other population group effects can be addressed by appropriate clinical trial designs, much more scientific information and research is needed in all areas of drug development to delineate the best possible approaches. Questions about the hormonal effects of drugs and pharmacokinetic and pharmacodynamic gender differences are not easy to resolve. An especially valuable area of focus might be whether in vitro studies in human tissues can be used predictively to rule in or rule out a possible gender effect. If successful, the capability of using these relatively low-cost approaches in a predictive way might ultimately reduce clinical trial costs. By learning more about gender effects, we will also be able to understand better about how medical therapies work, not only in population subsets, but also in the overall population. The need to clarify and refine basic principles of study design also becomes apparent as the best approaches to develop useful gender information are sought. Drawing on the deliberations of the 1995 Gender Workshop and working collaboratively with academia, industry, government, and other individuals and groups, FDA expects to identify further information and recommendations to assist sponsors in defining when a gender or other population group effects are clinically important.
Speakers List
Claudette Bain
HIV Law Project
Janice K. Bush, M.D.
Vice President
Janssen Research Foundation
Louis R. Cantilena, Ph.D., M.D.
Director, Division of Clinical Pharmacology University Services
University of the Health Services
Joy Cavagnaro, Ph.D.
Special Assistant to the Deputy Director
Center for Biological Research and Evaluation
Food and Drug Administration
Mei-Ling Chen, Ph.D.
Chief, Pharmacokinetics Evaluation Branch
Center for Drug Evaluation and Research
Food and Drug Administration
Neal Cutler, M.D.
Director, California Clinical Trials Medical Group
Paresh Dandona, B.Sc., M.B., B.S., F.R.C.P.
Professor of Medicine
State University of New York at Buffalo Chief of Endocrinology
Millard Fillmore Hospital
Jeanne DeJoseph, Ph.D., C.M.N
Department of Family Health School of Nursing
University of California at San Francisco
David Flockhart, M.D., Ph.D. Assistant Professor of Medicine and Pharmacology Georgetown Hospital
Victoria Hale, Ph.D.
Pharmacokinetics and Metabolism
Genentech
Jean Hamilton, M.D.
Director, Institute for Women's Health
Medical College of Pennsylvania and Hahnemann University
Eugene G. Hayunga, Ph.D. Office of Research on Women's Health
National Institutes of Health
David A. Kessler, M.D.
Commissioner of Food and Drugs
Food and Drug Administration
Sarah J.H. Kogut, M.S. Triology Consulting Company
Laura Kragie, M.D.
Critical Care Center
Center for Drug Evaluation and Research
Food and Drug Administration
Scott E. Lukas, Ph.D.
Associate Professor of Psychiatry Harvard Medical School
Murray K. Lumpkin, M.D.
Deputy Director, Review Management
Center for Drug Evaluation & Research
Food and Drug Administration
Stella Machado, Ph.D.
Chief, Research RAMPS and Methodology Planning Staff
Center for Drug Evaluation & Research
Food and Drug Administration
Theresa McGovern, J.D. HIV Law Project
Ruth Merkatz, R.N., Ph.D.
Director, Office of Women's Health
Food and Drug Administration
Robert O'Neill, M.D.
Director, Office of Epidemiology and Biostatistics
Center for Drug Evaluation & Research
Food and Drug Administration
Kimber Richter, M.D.
Deputy Director for Clinical and Review Policy
Center for Devices and Radiological Health
Food and Drug Administration
Jean Rowan, M.D.
Senior Director
Parke Davis
Janice Schwartz, M.D.
Northwestern University Medical School
Lewis Sheiner, M.D.
Professor of Laboratory Medicine University of California at San Francisco
Alan Sedman, M.D., Ph.D.
Parke Davis
Richard Simon, D.Se.
Chief, Biometric Research Branch National Cancer Institute
National Institutes of Health
Jean Louis Steimer, Ph.D.
F. Hoffmann-La Roche, Ltd.
Julie A. Swain, M.D.
Kenosha Hospital and Medical Center DeBakey Heart Institute
Robert Temple, M.D.
Associate Director for Medical Policy
Center Drug Evaluation & Research
Food and Drug Administration
Solomon Sobel, M.D.
Director, Division of Metabolic and Endocrine Drug Products
Center for Drug Evaluation & Research
Food and Drug Administration
Carol Trapnell, M.D.
Medical Researcher, Research Resources
Center for Drug Evaluation & Research
Food and Drug Administration
Edward Wallach, M.D.
Department of Obstetrics and Gynecology
Johns Hopkins University
Roger Williams, M.D.
Deputy Director for Science and Medical Affairs
Center for Drug Evaluation & Research
Food and Drug Administration
Janet Woodcock, M.D.
Director
Center for Drug Evaluation & Research
Food and Drug Administration
Raymond Woosley, M.D., Ph.D. Professor and Chairman
Department of Pharmacology
Georgetown University Medical Center
Return to Scientific Projects Page
Web page updated by clb 2001-JAN-30.