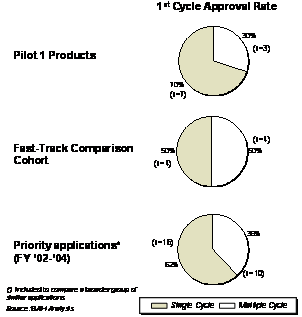|
 |
 |
FDA Home Page | Search
FDA Site | FDA A-Z Index | Contact
FDA | FDA Centennial
![]()
Independent Evaluation of the FDA's Continuous Marketing Application Pilot 1 and Pilot 2 – Initiatives & Evaluations
Contract No. 223-04-8100 Task No. T2
CMA Pilot 1 Evaluation and Pilot 2 Preliminary Evaluation Studies -- Final Report
May 10, 2006
![]()
TABLE OF CONTENTS
| 1 | |
| 6 | |
| 10 | |
| 10 | |
| 11 | |
| 13 | |
| 13 | |
| 14 | |
| 14 | |
| 18 | |
| 19 | |
| 20 | |
| 23 | |
| 23 | |
| 24 | |
| 29 | |
| 33 | |
| 33 | |
| 35 | |
| 5.3 Summary/Takeaways | 36 |
EXHIBITS
Exhibit 2–1. Comparison of the Fast-Track/Rolling Review and CMA Pilot 1 Programs |
7 |
Exhibit 2–2. Illustrative Differences between a Fast-Track IND and a CMA Pilot 2 Product |
8 |
Exhibit 2–3. Status of the Pilot 1, Pilot 2 and Comparison Cohort Products |
9 |
Exhibit 3–1. Evaluation Approach |
10 |
Exhibit 3–2. Hypothesis Generation Approach |
11 |
Exhibit 3–3. Data Sources and Level of Contribution |
12 |
Exhibit 4–1. Sponsor Motivations for Pilot 1 Participation |
13 |
Exhibit 4–2. First-Cycle Approval Rate for Pilot 1 Products, the Fast-Track/Rolling Review Comparison Cohort, and Priority NMEs (FY02-FY04) |
14 |
Exhibit 4–3. Products with Amendments Requiring Goal Extensions |
15 |
Exhibit 4–4. Number of Issues Identified for Pilot Products Versus the Comparison Cohort As An Indicator of Application Quality |
16 |
Exhibit 4–5. FDA Met Pilot 1 Six-month Feedback Expectations |
17 |
Exhibit 4–6. Average Number of Meetings and Communications for Pilot 1 Products Versus the Comparison Cohort |
17 |
Exhibit 4–7. Reviewable Unit Interdependencies |
18 |
Exhibit 4–8. Limitations of Submitting Early Reviewable Units |
19 |
Exhibit 4–9. Timing of Reviewable Units in Relation to the Complete Submission of the Pilot 1 NDA/BLA |
20 |
Exhibit 4–10. Factors Beyond the Pilot 1 Process Effecting First-cycle Review Outcome |
21 |
Exhibit 4–11. Multi-cycle Pilot 1 Products Would Have Had The Same Outcome Regardless of Pilot Status |
21 |
Exhibit 4–12. Some Fast-Track/Rolling Products Received Early Review and Feedback |
22 |
Exhibit 4–13. Fast-Track/Rolling Products With Limited Evidence of Early Review |
23 |
Exhibit 4–14. Major Fast-Track Activities Versus Additional Pilot-specific Activities Reported by Participating Sponsors |
24 |
Exhibit 4–15. Major FDA Fast-Track Review Activities Versus Additional Incremental Pilot 1-specific Activities |
26 |
Exhibit 4–16. Incremental Effort for FDA of Pilot-Specific Activities per Application (1 cycle) |
27 |
Exhibit 4–17. Incremental Effort of Pilot-Specific Activities by Function (1 cycle) |
27 |
Exhibit 4–18. Extrapolation of the Incremental Pilot Effort to the Average Number of Fast-Track Products Submitted to Divisions with Pilot 1 Experience |
28 |
Exhibit 4–19. Three Year Total Fast-Track Submissions by Divisions (FY2003-2005) |
29 |
Exhibit 4–20. Scenarios on Potential FDA Re-Work |
30 |
Exhibit 5–1. Details of the Meeting Schedule Agreement – Fixed vs. Trigger |
34 |
Exhibit 5–2. Analysis of the Fixed Schedule Method – Communication Increase and FDA Time Effort |
35 |
Exhibit 5–3. Sponsor Motivations for Program Participation and Perceived Benefits |
36 |
| Exhibit 5–4. Early Observations on the Pilot 2 Program | 37 |
This report summarizes the results of an evaluation of the Food and Drug Administration's (FDA) Continuous Marketing Application (CMA) Pilots 1 & 2. The Pilots were part of the Prescription Drug User Fee Act (PDUFA) reauthorized in 2002, also known as PDUFA III.
The evaluation included interviews of FDA and sponsors who participated in the Pilots, and also included interviews with several sponsors who had eligible products, but opted not to participate. The group of non-pilot, eligible products formed the basis for a comparison cohort (i.e., Fast-Track/Rolling Review products submitted between 2002-2004) against which the Pilots were measured to determine any differences. In addition to interviews, the evaluation incorporated available FDA data sources such as product action packages, FDA's time tracking system, PDUFA goal date tracking system, and document filing system. All collected data were then used to evaluate the impact, benefit and effort of the Pilots.
CMA Pilot 1
CMA Pilot 1 allowed eligible sponsors submitting a New Drug Application (NDA) or Biologics License Agreement (BLA) to submit Reviewable Units (RUs)-early submissions of complete sections of the application. FDA committed to review the RUs within 6 months and provide the sponsor with a discipline review letter upon completion of the review.
Pilot 1 products were compared to similar non-pilot products to determine any differences in program outcomes, including first cycle review rates, application quality (i.e., based on the number of issues reported during the review and the number of amendments requiring extensions), and the number of FDA/sponsor communications. Other factors were examined including the impact of RU timing and order, RU interdependencies, the impact of early review, and sponsor's motivations for participating in the Pilot. Also, the evaluation analyzed the additional incremental effort of the Pilot for sponsors and FDA over any other non-Pilot, Fast Track/Rolling reviewed product.
A brief summary of the Pilot 1 program outcome findings are as follows:
Sponsor Motivations: FDA's commitment to a 6-month review of the reviewable unit submissions was a primary motivator for eligible sponsors to participate in the Pilot.
1st-cycle Approval Rate: Pilot 1 products had a favorable 1st-cycle approval rate (70%). The Pilot product approval rate, however, was not significantly different from the historical approval rate for priority-reviewed products between 2002-2004 (i.e., product receiving a 6-month review).
Application quality: Two metrics identified as proxy indicators of application quality for purposes of this evaluation were the number of issues identified by FDA in the application and the number of amendments requiring extensions on the PDUFA goal date. The results of these metrics for the Pilot products and the comparison cohort were similar-indicating that the Pilot did not appear to have any impact on application quality.
FDA/sponsor communications: There was no significant difference between the number of FDA/sponsor communications for the Pilot 1 products and for the comparison cohort products. .
RU Interdependence: RU Interdependence was evaluated to determine if segmented early RU submissions impacted the review. FDA and sponsors indicated that Chemistry, Manufacturing and Control (CMC) is considered the most independent RU, and therefore the one that would potentially benefit the most from early submission. On the other hand, development of the Clinical section is often the rate-determining step, and thus typically submitted last with the complete application submission.
RU Timing: Few sponsors can have a complete RU submission 12 months prior to the complete submission. For CMC, most sponsors considered 3 to 6 months prior to the complete submission the more feasible RU submission timeframe. These limitations on early submission prevented maximizing the opportunity for early review and subsequent feedback in the Pilot.
Early Review: One Pilot 1 product benefited from early review. Other Pilot products with 1st-cycle approvals would have likely had favorable actions regardless of early review because it seemed that the unmet medical need nature of the products had influence on the risk/benefit analysis and the high level of attention the application received. Also, for some of the products, a major issue may have been identified in the sections submitted with the complete application, therefore, although many of those issues were addressed within the first cycle, there was no benefit of early review or feedback.
Effort: The evaluation also included an analysis of the additional effort for sponsors and FDA to participate in the Pilot. Sponsors indicated that the incremental effort for Pilot 1 was minimal. Aside from completing and submitting the Pilot application, they reported that the activities they conducted were similar to any application submission. In addition, smaller-sized sponsors indicated that the Pilot helped distribute their workload. On the other hand, FDA incurred the majority of the effort in launching the Pilot at the Division level. Most of the FDA participants reported that incremental on-going efforts of the Pilot were not overly burdensome, with Regulatory Project Managers incurring the highest effort increase compared to others on the review team. Based on the effort data collected, FDA's incremental effort for Pilot 1 was estimated to be between 190-360 direct labor hours per application. This incremental effort was above the effort that would have been expended for a non-pilot, Fast-Track, priority review in the Divisions that experienced a Pilot 1 review.
The result of the evaluation was that there is no conclusive finding that indicates whether the Pilot 1 program should continue or be terminated. This may be due to several influencing factors such as the small sample size of the Pilot and the comparison cohort and the high unmet medical need nature of many of the products. The key findings of the evaluation showed:
- Sponsors valued FDA's 6-month RU review commitment
- Helped distribute sponsors' workload
- Additional time to address issues for early submitted RUs is a review process benefit
- A strong first-cycle approval rate
- Similar level of application quality
- Similar levels of communication.
Given that this evaluation focused on the comparison of the Pilot 1 program to the Fast Track/Rolling Review program, many Pilot participants offered their perceptions of the Fast Track program. Industry strongly valued the subtle differences that Pilot 1 offered over the Fast Track/Rolling Review program where FDA remained neutral.
While this analysis uncovered no resounding reason to continue the Pilot as a separate program, there may be merit to integrating some positive attributes and lessons-learned from the Pilot 1 program into the existing Fast-Track/Rolling Review structure. For example, some challenges with the current Fast Track/Rolling Review program are:
For these particular challenges, the Pilot 1 structure offers potential improvements over the Fast Track/Rolling Review program that include:
If the Fast Track/Rolling Review program were modified with these Pilot 1 attributes, this would allow the FDA to plan better for reviews because they can expect a complete section for early review; early review would be conducted consistently across FDA divisions for early submissions; and issues would be identified earlier, and in some cases, may lead to resolution prior to the first action date, or may help reduce the time between cycles if sponsors can begin addressing deficiencies earlier.
Some considerations before deciding to make any modifications may include:
Additionally, if modifications are implemented, other considerations may include:
Further, if the FDA decides to implement any modifications, additional resources would be required since the Agency would incur most of the additional workload burden. If implemented, the FDA may incur, in addition to the incremental costs described in this report, additional costs during the transition phase as this program is rolled out more broadly to the Divisions which in parallel need to complete reviews of other applications currently under review. It is imperative that the FDA receive additional resources commensurate with the effort incurred to transition to and maintain the new process, in order to ensure that review Divisions are not overburdened.
CMA Pilot 2
CMA Pilot 2 allowed eligible sponsors to establish an agreement with FDA to have scientific exchanges (e.g., meetings, protocol reviews, document reviews) throughout the product development process. This evaluation for Pilot 2 is preliminary. Most of the products are still in the middle of product development, so it is too early in the process to analyze the impact of the program outcome. Therefore, the evaluation focused on interim FDA and sponsor perceptions of the Pilot through interviews and FDA's data systems mentioned above.
Findings showed that there were two different Pilot 2 approaches sponsors used to schedule exchanges with FDA-one approach established an estimated schedule in advance (the Fixed Schedule) and the other focused on when FDA could provide feedback based on the type of interaction (the Trigger Method), scheduling interactions as needed. Due to the uncertainty of product development, the schedules established early using the Fixed Schedule approach often slipped. The Trigger Method was received more positively from both FDA and the sponsors. Sponsors received guaranteed feedback that they valued the most from the Pilot, and the FDA did not feel committed to a blanket agreement to grant all interactions before their need was determined. Also, the negotiation process between the sponsor and FDA for the Trigger Method approach was much less burdensome than the process for agreeing to a fixed schedule of meetings.
In terms of effort, following the Fixed Schedule method and actually conducting all planned meetings, could result in a doubling of the number of communications for a Pilot 2 product over the Fast-Track program. The estimated incremental effort is over 500 hours for a Pilot 2 product over a Fast-Track product, based on a Fixed Schedule approach.
Although it is too early in the Pilot 2 program to determine the value of its impact, there were early observations/takeaways to take into account:
In 1992, Congress passed the Prescription Drug User Fee Act (PDUFA) authorizing the FDA to collect fees from companies for the review of drug applications as well as for providing regulatory oversight of manufacturing plants. Congressional funds and industry user fees, established under the PDUFA legislation, fund additional resources that allow the FDA to meet drug-review performance goals. In accordance with the 2002 PDUFA Reauthorization Performance Goals and Procedures (PDUFA goals), the FDA agreed to meet specific performance goals to improve the effectiveness and efficiency of FDA review of New Drug Applications (NDAs) and Biologic Licensing Applications (BLAs).
Overview of Continuous Marketing Application Pilots 1 & 2
As part of the current Fast-Track program, the FDA allows for the submission of Rolling Reviews-pre-submitted portions of marketing applications. There is however, no commitment for early review, which depends on resource availability. When reviewers are available, deficiencies of early-submitted sections can be identified earlier, providing in some instances an opportunity for resolution prior to first review action, and ultimately reducing the time for important drugs to reach the market.
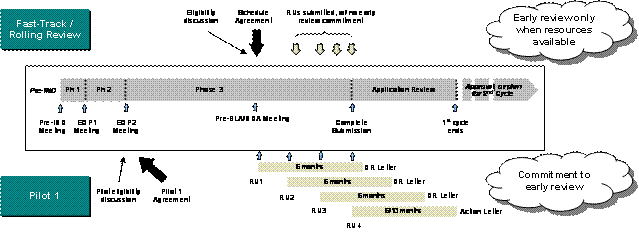
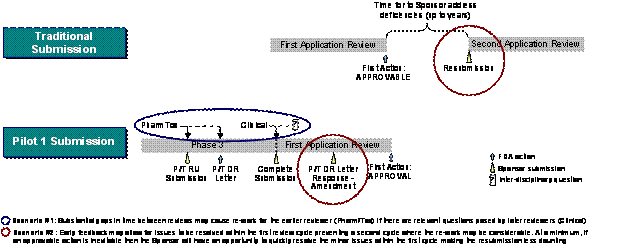
| The Continuous Marketing Application (CMA) Pilot 1 program, introduced in PDUFA III, formalizes the commitment for early review of complete sections of marketing applications. Eligible Fast-Track applications can be submitted in up to four pre-determined sections called Reviewable Units (RUs) within one year of complete application submission. Under this Pilot, the FDA commits to reviewing these pre-submitted portions and issuing a discipline review letter within six months of receipt of each RU. In addition to the early feedback commitment, other goals of Pilot 1 included increasing review efficiency (e.g., eliminate the need for resubmission and multiple review cycles) and possibly reducing time to market (e.g., focused RU submissions may increase application quality; early issue resolution may help increase first cycle approvals). Exhibit 2–1 illustrates the differences in Pilot 1 to a traditional Fast-Track application. | General CMA Pilot 1 program criteria:
|
Exhibit 2–1. Comparison of the Fast-Track/Rolling Review and CMA Pilot 1 Programs
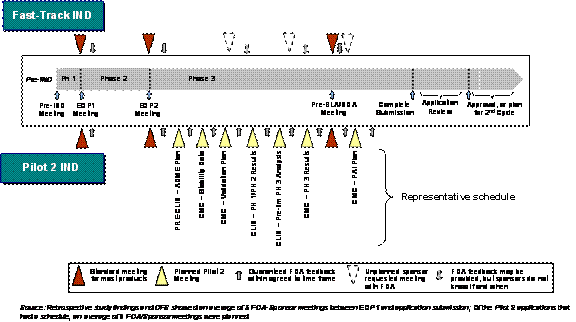
A second CMA Pilot was also established under the PDUFA III legislation, which allows applicants with eligible Fast-Track products to enter into an agreement with the FDA that ensures frequent scientific feedback and interaction during the investigational new drug (IND) phase of product development. The intent of the early interaction is to ensure that the FDA's and sponsors' expectations for the clinical development program are aligned, preventing the need for re-work, and eliminating unnecessary trials. To be eligible, applicants must have a Fast-Track designated product, engage with the FDA at an End-of-Phase 1, or equivalent meeting, and demonstrate that the product has the potential to significantly benefit the public health. Applicants are required to draft an agreement for proposed feedback and interactions with FDA, defining the timing and frequency of FDA-sponsor contacts, the general types of submissions that will stimulate feedback, and the forms of communication requested. Exhibit 2–2 provides an overview of the key differences between a Fast-Track versus a CMA Pilot 2 product. |
General CMA Pilot 2 program criteria: |
Exhibit 2–2. Illustrative Differences between a Fast-Track IND and a CMA Pilot 2 Product
CMA Pilot Evaluation and Report
Under PDUFA III, the FDA agreed to retain an independent, expert consultant to evaluate the value and costs of the CMA Pilots 1 and 2, both from the perspective of the FDA and participating sponsors. This report presents the full evaluation of CMA Pilot 1 and a preliminary evaluation of CMA Pilot 2 which is currently still in early stages.
The scope of the evaluation included 11 Pilot 1 and 9 Pilot 2 products (see Overview of CMA Pilots 1 & 2). A product comparison cohort was selected from the Fast-Track products submitted between 2002-2004 that were not enrolled in the Pilot programs. In that time frame, there were 12 non-pilot, Fast-Track products, and of those products 8 were selected as the comparison cohort. The 8 products were selected because they had Rolling Review submissions (i.e., portions of the application submitted prior to the submission of the full NDA/BLA) and did not have a special designation (e.g., 505(b)(2)), to ensure the comparison cohort was similar to the eligible Pilot products. Exhibit 2–3 below depicts details of the Pilot 1, Pilot 2 and comparison cohort products as of January 10, 2006. All comparison products have reached first action; 10 of the 11 Pilot 1 products reached first action, with one sponsor withdrawing after Pilot program enrollment. Because Pilot 2 targets products that are in earlier phases of development, only one of the products in this cohort has advanced to the stage of application submission. |
Comparison cohort selection criteria included: |
Exhibit 2–3. Status of the Pilot 1, Pilot 2 and Comparison Cohort Products
Pilot 1 |
Status |
Pilot 2 |
Status |
Comparison Cohort |
Status |
|---|---|---|---|---|---|
Retisert |
Approved |
8 Products |
IND |
Aldurazyme |
Approved |
Tarceva |
Approved |
1 Product |
NDA |
Apokyn |
Approved |
Macugen |
Approved |
|
|
Iressa |
Approved |
Exjade |
Approved |
|
|
Alimta |
Approved |
Kepivance |
Approved |
|
|
Fuzeon |
Approved |
Orencia |
Approved |
|
|
Velcade |
Approved |
Nexavar |
Approved |
|
|
2 Products |
Approvable |
2 Products |
Approvable |
|
|
|
|
1 Product |
Not Approvable |
|
|
|
|
1 Product |
Withdrawn |
|
|
|
|
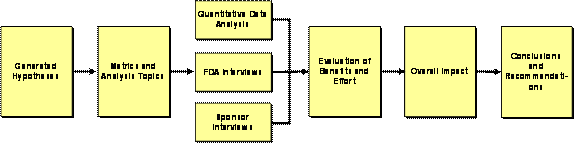
The approach for the evaluation was based on a series of hypotheses formulated at the beginning of the task to capture costs and benefits to the FDA reviewers and sponsors. Metrics were selected based on these hypotheses to direct data collection and ensure focused interview questions. As themes emerged, hypotheses were narrowed to those that warranted further research/analysis. From analyses, an assessment of the overall impact was determined which led to final conclusions and recommendations. Exhibit 3–1 summarizes the overall evaluation approach.
Exhibit 3–1. Evaluation Approach
Hypotheses developed to evaluate both Pilots focused on benefits in terms of value and meeting FDA strategic goals (e.g., decreasing time to approval) and effort/cost to the FDA and sponsors. Metrics derived from hypotheses captured both quantitative and qualitative aspects (see section 3.2). Examples of measures used for the evaluation included: first-cycle approval rate, number of amendments submitted per application, perceived workload distribution, and quality of submissions.
Examples of hypotheses are provided in Exhibit 3–2.
Exhibit 3–2. Hypothesis Generation Approach
Evaluation Factors |
Key Hypothesis Areas |
Sample Hypotheses |
|---|---|---|
Contributing Factors |
• RU interdependence • Issue Resolution • Application Quality • Communication • Sponsor Characteristics • Sponsor Motivations |
• Sponsors have more time to resolve issues prior to first action • Pilot 1 is more effective for large and US-based sponsors • Pilot 1 benefits some therapeutic areas more than others • RUs cannot be reviewed independently |
Benefits |
• PDUFA Goal • Time to Approval • First-Cycle Approval
|
• FDA meets goal for RUs reviewed w/I 6 months • Pilot 1 applications have hogher single approval rate • Pilot 1 Priority reviews are more likely to be completed on time • Pilot 1 applications require fewer extensions |
Effort/Costs |
• Resource Allocation • Workload Distribution • Incremental Effort (Start-Up, Recurring)
|
• Pilot 1 will require a large effort to implement • Workload for Pilot 1 applications is distributed more evenly • Pilot 1 will displace other FDA activities |
Quantitative sources included:
Although these were important sources of data, there were some limitations including:
As a result, the evaluation also required qualitative information collected through interviews with FDA review teams and Sponsors (Exhibit 3–3).
Exhibit 3–3. Data Sources and Level of Contribution
Data from various information sources contributed significantly to the CMA Pilot
evaluations and FDA and sponsor interviews were a major contribution .
Data Source |
Pilot 1 |
Pilot 2 |
|---|---|---|
Action Packages |
7 out of 11 |
N/A |
Discipline review Letters |
10 out of 11 Products |
N/A |
Time-tracking data |
Limited Pilot data |
Limited Pilot data |
Meeting tracking data (IMTS) |
Mtg timelines available |
Mtg timelines available |
Pilot Status tracking data |
Status data available |
Status data available |
Division File Ststem (DFS) |
Meeting minutes, etc |
Meeting minutes, etc |
Sponsor Pilot 2 Agreements |
N/A |
Planned meetings |
FDA Interviews |
Pilot 1 |
Pilot 2 |
|---|---|---|
Office/Deputy Office Director |
2 |
2 |
Div. Dir./Review Team Leaders |
7 |
3 |
Discipline Reviewers |
7 |
0 |
Regulatory Project Managers |
10 |
8 |
Sponsor Interviews |
Pilot 1 |
Pilot 2 |
|---|---|---|
Pilot 1 Sponsors |
8 out of 11 |
N/A |
Pilot 2 Sponsors |
N/A |
7 out of 9 |
Comparison Cohort Sponsors |
5 |
5 |
Since introducing the Pilot 1 program in October 2003, FDA accepted 11 applications. This relatively small number of products does not allow in-depth statistical analyses to substantiate findings. Thus, the evaluation and corresponding recommendations are based on both the quantitative results and qualitative observations of FDA review participants, sponsor Pilot participants, and several sponsors eligible to participate, but who chose not to apply or participate.
The key findings of the Pilot are presented in the following four sections:
3.1 Sponsors Motivations for Participation-Presents a summary of the sponsor motivations and program expectations based on interview findings
3.2 Impact-Discusses the potential benefits (first-cycle approval rate, amendments triggering extensions, number of communications, etc.) of the program that were evaluated quantitatively and qualitatively
3.3 Incremental Effort for Sponsors and FDA-Presents the evaluation of the incremental effort to implement the Pilot
3.4 Summary/Recommendations-Discusses the findings and the reasoning behind the final recommendations
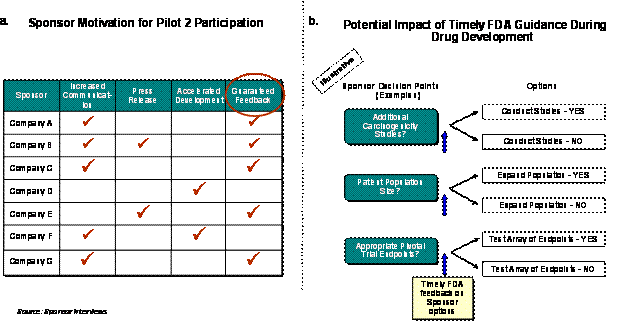
FDA's commitment to an earlier review with a 6-month review clock was the primary motivator for companies to participate. Additionally, sponsors cited a number of desired outcomes that made the Pilot opportunity attractive. These were mostly related to the potential for increased communication and publicity, and the prospect of expedited review beyond a non-Pilot review (Exhibit 4–1). In contrast, sponsors that were eligible for Pilot 1 enrollment but chose not to participate, noted that current access to the FDA under the Fast-Track/Rolling Review process was satisfactory and therefore found no additional incentives to enroll.
Exhibit 4–1. Sponsor Motivations for Pilot 1 Participation
The themes that emerged during the evaluation are addressed in the following four areas:
Meeting the specific Pilot expectations/goals was a key evaluation factor. The analysis examined first-cycle approval rates, quality of applications, number of deficiencies and effectiveness of communications. The first-cycle approval rate was of high interest since it is a highlighted PDUFA III goal as a means to ensure earlier access of new promising products that address a high unmet medical need. Because of the Pilot 1 eligibility criteria, a majority of the products enrolled fit into this high unmet medical needs category.
Of the 11 Pilot products, one was withdrawn by the sponsor due to lack of efficacy shortly after the first RU was submitted. As a result, only 10 products were included in the analyses. Seven of the 10 Pilot 1 products received first-cycle approval (70%), compared to a 50% first-cycle approval rate comparison cohort products. The Pilot result is similar to the first-cycle approval rate observed for New Molecular Entities (NMEs) submitted between 2002-2004, that received priority review (62%). Because of the low number of products in the Pilot and the comparison cohort, the larger comparison group of priority reviewed NMEs addressing high unmet medical needs were included to provide an additional perspective on the first-cycle approval trends for Pilot 1 products (Exhibit 4–2).
Exhibit 4–2. First-Cycle Approval Rate for Pilot 1 Products, the Fast-Track/Rolling Review Comparison Cohort, and Priority NMEs (FY02-FY04)
While the first-cycle approval results are not materially higher for Pilot 1 products, it is important to note that the 3 products that did not receive a first-cycle approval failed to demonstrate efficacy. Therefore, an early review would not be expected to provide significant benefit given the long lead times necessary to plan and conduct additional studies - a frequent requirement for such deficiencies. In contrast, analyzing multiple cycle priority applications (Exhibit 4-2) from a previous study [1], factors influencing multiple cycles included: application quality, CMC and safety issues. These deficiencies were either not observed and/or were adequately addressed in the case of Pilot 1 products with first-cycle approvals. While this may imply that the Pilot's early review structure has a positive influence, the impact of the small sample size remains unknown.
In addition to first-cycle approvals, the Pilot 1 evaluation also analyzed the impact on application quality. Using proxy indicators, quality was assessed through:
Applications that did not require amendments were considered complete and hence of higher quality compared to submissions that involved amendments. Similarly, fewer number of communications, for example to discuss deficiencies and FDA requests for information, was used as a proxy indicator of higher quality submissions.
Two of the 10 Pilot 1 products had amendments that required goal extensions. In these cases, the amendments requiring extensions were triggered by FDA requests for further information that would have occurred regardless of Pilot status (i.e., they were not the result of a discipline review letter). This is consistent with the comparison cohort and the broader comparison group of priority NMEs, suggesting that there is no change in application quality (Exhibit 4–3).
Exhibit 4–3. Products with Amendments Requiring Goal Extensions
Furthermore, the Pilot did not have an impact in terms of number of deficiencies, requests for information from FDA to sponsors, and other clarification communications (Exhibit 4–4). This may suggest that application quality neither increased nor decreased based on what could be observed. Moreover, FDA interviews further confirmed that they did not perceive an impact on quality for Pilot 1 applications.
Exhibit 4–4. Number of Issues Identified for Pilot Products Versus the Comparison Cohort As An Indicator of Application Quality
From the sponsor's perspective, a key outcome/expectation was the 6-month feedback for each RU submitted. The FDA was able to meet the 6-month review commitment for early submissions for all Pilot 1 products (Exhibit 4–5).
Exhibit 4–5. FDA Met Pilot 1 Six-month Feedback Expectations
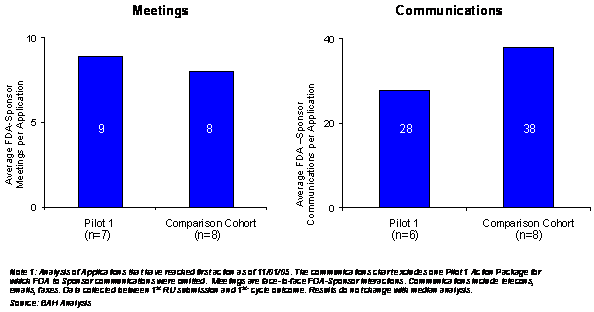
The secondary desired outcome expressed by sponsors, was to have increased communication with FDA during the Pilot 1 process. As Exhibit 4–6 displays, in comparison to other Fast-Track/Rolling Review products, Pilot 1 products appear to have similar number of communications with FDA.
Exhibit 4–6. Average Number of Meetings and Communications for Pilot 1 Products Versus the Comparison Cohort
At the onset of the Pilot 1 program, a concern for both FDA and sponsors was the impact of a segmented review, resulting in incomplete submissions or the need to repeat analysis of previously reviewed sections. Therefore, the evaluation examined the perceptions of the interdependencies of the RUs for both FDA and sponsors, for example:
In the case of the ten Pilot 1 applications, the four RUs were typically:
In general, both FDA and sponsors agreed that CMC was the most independent RU and should be submitted as one of the first RUs. The Clinical section on the other hand was broadly considered to be rate-limiting in most product development programs, and relying most heavily on other sections of an application. Exhibit 4–7 summarizes the FDA and sponsor interview findings in regards to the interdependence of the RUs.
Exhibit 4–7. Reviewable Unit Interdependencies
The Reviewable Unit Interdependencies are rated on a scale of "0" for "Not
Interdependent" to "4" for "Very Interdependent".
|
Clinical |
Pharm/Tox |
BioPharm |
Comments |
|---|---|---|---|---|
Clinical |
N/A |
2 |
4 |
• Clinical incorporates final label inputs from all disciplines • Availability of clinical data is the rate determining step in most development programs |
Pharm/Tox |
2 |
N/A |
2 |
• Pharm/Tox often interacts with Clinical and BioPharm; however, Pharm/Tox is commonly ready to submit first |
BioPharm |
4 |
2 |
N/A |
• BioPharm interacts heavily with Clinical and is often submitted at the same time |
CMC |
1 |
1 |
1 |
• The CMC section would benefit most from early submission (e.g., manufacturing inspections could be scheduled earlier, especially if in a foreign country) |
While early CMC submission was desirable, most sponsors expressed several practical constraints:
These considerations are further magnified in the case of biologics due to the relatively higher manufacturing costs, need for validation lots and often shorter shelf-lives. The consensus among sponsors was that in general, CMC sections could not be prepared significantly earlier than 3-6 months prior to the rest of the submission.
Similar practical constraints were also noted for other sections, implying that the 12 month submission window between first and last sections of an application can rarely be fully leveraged (Exhibit 4–8).
Exhibit 4–8. Limitations of Submitting Early Reviewable Units
CMC |
• Submitted as late as possible for shelf-life considerations and sale concerns • Stability data often not ready more than 3 months prior to full submission • Biologics: More validation lots, shorter shelf-life (stability), global shortage of bioprocess capacity, difficult to optimize full-scale production |
|---|---|
Clinical |
• Most interdependent section, rate limiting • Early clinical results may become out-dated, submission/resubmission of updated information may trigger extension |
Preclinical |
• Decision to conduct expensive carcinogenicity studies are generally started as late as possible to mitigate costs |
Of the Pilot 1 products, only one sponsor was able to submit a RU greater than 6 months prior to the complete submission. More than half of the participating companies submitted the first RU only within 3-6 months prior to the complete submission, and only a few of the companies submitted their first RUs within 0-3 months of the complete submission. Sponsors noted that the uncertainty of product development and the challenges of preparing a comprehensive RU too far in advance of the complete submission impacted RU submission timing. In addition, FDA's CMA Pilot guidance specified that no more than four RUs would be accepted per application. One sponsor submitted four RUs-a majority submitted between two to three RUs (Exhibit 4–9).
Exhibit 4–9. Timing of Reviewable Units in Relation to the Complete Submission of the Pilot 1 NDA/BLA
| Product | Timeline of RU Submissions Prior to Complete Submission (Mos) | # of RUs | Time First-Last RU (mos) |
|---|---|---|---|
| A | 12 & 8 | 2 | 12 |
| B | 4.5 & 3 | 2 | 4.5 |
| C | 4.5 | 1 | 4.5 |
| D | 4.5, 4 (2 RUs), &1.5 | 4 | 4.5 |
| E | 3.5 | 1 | 3.5 |
| F | 2 & 1 | 2 | 2 |
| G | 3 (3 RUs) | 3 | 3 |
| H | 1 (2 RUs) | 2 | 1 |
| I | 1 | 1 | 1 |
| J | 2.5 & .5 (2 RUs) | 3 | 2.5 |
Although there seems to be a positive trend in first-cycle approvals for Pilot 1 products versus the comparison cohort (Exhibit 4–2), this may not be solely due to the review of early submitted portions of an application and subsequent feedback. Some other factors may come to play that contribute to the first-cycle approval include:
A review of Pilot 1 product outcomes suggests that other factors beyond early review may have contributed to favorable first-cycle outcomes, irrespective of Pilot participation. The early RU review for Product C for example, had no critical deficiencies that could have led to a delay in approval (Exhibit 4–10). In the case of Product G, the majority of deficiencies were identified with the complete submission and not in the early submitted RUs (Exhibit 4–10). Of the products analyzed, it appears that only Product E benefited significantly from early CMC RU review, which increased the time available for the sponsor to address the issues within the first-cycle (Exhibit 4–10).
Exhibit 4–10. Factors Beyond the Pilot 1 Process Effecting First-cycle Review Outcome
Product |
Pilot 1 Benefit? |
Comment |
|---|---|---|
B |
no |
• CMC delayed and filed as final RU - several issues were identified requiring an amendment, and triggering a 3-month goal extension |
C |
no |
• Only minor issues were discussed during the review; did not provide a basis for an approval delay |
E |
yes |
• One RU, CMC, was submitted four months before the complete submission; early submission allowed CMC issues to be addressed easily |
G |
no |
• Major issue in CMC discipline which was the RU submitted - there was no added benefit from the pilot |
H |
no |
• Product addresses high unmet need; would have received high attention regardless of filing/review designation |
I |
no |
• CMC issues elevated a little sooner due to early RU; timing of other issues, e.g., immunogenicity, was similar to a regularly filed priority review |
J |
no |
• Product addresses high unmet need; would have received high attention regardless of filing/review designation |
The outcome of three Pilot 1 products that did not receive first-cycle approval is likely to also be independent of the review process, since all three failed to adequately demonstrate efficacy: a deficiency that typically requires significantly longer resolution time than afforded by early feedback under Pilot 1 (Exhibit 4–11).
Exhibit 4–11. Multi-cycle Pilot 1 Products Would Have Had The Same Outcome Regardless of Pilot Status
Product |
Pilot 1 Benefit? |
Comment |
|---|---|---|
A |
no |
Failed to meet non-inferiority criterion for primary efficacy; new study required |
D |
no |
Failed to demonstrate efficacy due to an unacceptable study population; not approvable |
F |
no |
Failed to meet clinically meaningful endpoints; new study required |
In comparison, several of the Fast-Track/Rolling Review products that received early review and feedback prior to the complete submission, as identified by a FDA request for information, also received 1st-cycle approval. Similar to the Pilot products however, additional factors may have influenced the outcome, such as the unmet medical need (Exhibit 4–12).
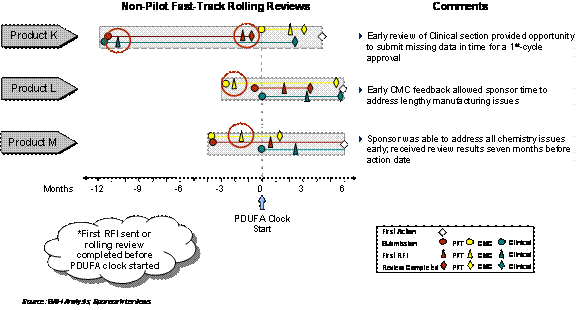
Exhibit 4–12. Some Fast-Track/Rolling Products Received Early Review and Feedback
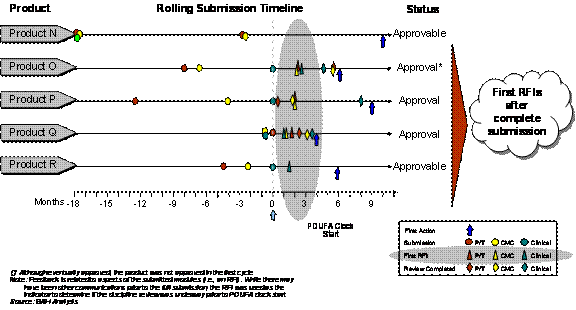
In general however, it is not clear whether an early review of Rolling Submissions occurs consistently. Using FDA's request for information (RFI) as an indicator, 3 of 8 Fast-Track applications that submitted rolling portions of the application received an early review (Exhibit 4–12). For the other 5 applications, it is difficult to ascertain whether or not a review began before the complete submission since the first RFI was not communicated until after the complete submission (Exhibit 4–13).
As with the Pilot products, the comparison cohort shows that the impact of early review on first-cycle approvals is still unclear. Of the 4 comparison cohort products that received first cycle approval, only 2 received early review. This again demonstrates that factors in addition to or independent of early review and subsequent feedback contribute to a favorable first-cycle outcome.
Exhibit 4–13. Fast-Track/Rolling Products With Limited Evidence of Early Review
This section analyzes the incremental effort to implement Pilot 1 for sponsors and FDA. The incremental effort is related to activities specifically conducted to prepare and review products under the Pilot program above what would typically be expended for a Fast-Track/Rolling review product The assessment of these additional activities was the basis for estimating the incremental effort related directly to the Pilot.
Sponsors that participated in the Pilot program noted that the majority of activities related to application preparation and submission would have been performed irrespective of the submission route. Only few additional activities were reported with minimal incremental effort (Exhibit 4–14). These were comprised of:
Exhibit 4–14. Major Fast-Track Activities Versus Additional Pilot-specific Activities Reported by Participating Sponsors
Major Sponsor Fast Track Activities
Pre-submission |
Post-submission |
|---|---|
Request for Fast-Track Designation |
Respond to FDA RFIs |
EOP1 Meeting |
Submit Amendments (if applicable) |
EOP2 Meeting |
Receive Action Letter (at 6-10 months) |
SPA |
|
Pre-NDA/BLA Meeting |
|
Prepare Application Sections |
|
Submit Complete Sections |
|
Additional Sponsor Pilot-Specific Activities
Pilot-specific Activities |
Incremental Effort (# Labor Hours) |
Comment |
|---|---|---|
Pilot Application Preparation |
<100 |
Sponsors reported the Pilot 1 application effort was minimal |
Internal Meetings |
<100 |
Sponsors reported 1-3 additional meetings |
Meetings with FDA |
N/A |
Sponsors reported no Pilot-specific meetings with FDA |
Furthermore, many sponsors mentioned that the Pilot helped with internal workload distribution, a benefit especially highlighted by smaller companies with limited resources dedicated to application preparation and interfacing with the FDA review team. Specifically, sponsors noted some pre-submission and post-submission workload benefits resulting from the Pilot:
Overall, the sponsors perception of Pilot 1 was that the effort involved was not burdensome and there was no downside to participating in the program.
Similar to the effort analysis conducted for sponsors, the Pilot effort for FDA was measured in terms of additional activities that FDA reviewers needed to perform beyond what is typically necessary for the Fast-Track/Rolling reviews. The activities included start-up efforts, additional filing meetings, Pilot admittance/RU schedule negotiations, acknowledgement/discipline review letter writing and review, additional internal FDA meetings, and RU re-work. The additional Pilot-specific activities for FDA are described below and are also summarized in Exhibit 4–15.
Start-Up Effort
As this was a new program, each
Division receiving a Pilot 1 product incurred start-up efforts associated with
establishing how to approach the reviews, for example, by creating appropriate letter
templates and/or ensuring the review team were aware of the requirements of the
Pilot for review purposes. By design, the CMA guidance did not specify Pilot
implementation, therefore, each Division addressed this separately. Divisions
that had multiple Pilot 1 products noted that this effort was mostly incurred
with the first Pilot submission. Subsequent Pilot products accepted into the
same review Division required minimal additional startup effort.
Additional Filing
Meetings
In a typical Fast-Track/Rolling
review, FDA review Divisions will hold one filing meeting after the complete
submission. Under the Pilot, two of the Divisions interviewed held a filing
meeting for each RU in addition to a filing meeting after the complete
submission. Other Divisions continued to hold one filing meeting upon receipt
of the complete submission; however, a determination whether each RU is
substantially complete and meets the conditions of the Agency agreement within
60 days must still occur.
Pilot Admittance/RU
Schedule Negotiations
FDA incurred costs for reviewing
each request for Pilot participation in order to determine eligibility. For
products that were ultimately accepted, some review team members reported minimal
negotiation time with sponsors regarding the most appropriate RU schedule.
Acknowledgement/Discipline
Review Letters
The FDA incurred additional
costs for letters acknowledging the receipt of RUs or reporting the results of
discipline reviews. These included direct costs associated with developing the
communications as well as multiple levels of review.
Internal FDA Meetings
Aside from start-up meetings
to ensure that the review team was aware of the review process under Pilot 1,
there were not many additional Pilot-specific meetings reported. However,
there were a few instances where a typical internal meeting that may only
happen once during the course of a non-Pilot review, happened two or more times
due to the review of multiple RUs plus the complete submission under the Pilot.
RU Re-work
A concern voiced by FDA
reviewers was the potential for duplicative review efforts-initially upon
receipt of the RU, and again during review of later sections that required a
reference to prior RUs. Alternatively, certain deficiencies identified upon
early RU review be resolved by the sponsor in time for re-submission during the
first action. In practice however, there was only one product where some
incremental RU rework was required. Most reviewers noted that RU reviews did
not differ in terms of level of effort or approach compared to any product
review with a 6-month clock. In particular, it was noted that RUs were
typically not reviewed more than once. Two of the Pilot products had amendments
to RUs that triggered clock extensions, but these amendments likely would have
occurred regardless of Pilot status. Therefore, the re-review work on the
amendments was not viewed as a Pilot-specific incremental effort.
Exhibit 4–15. Major FDA Fast-Track Review Activities Versus Additional Incremental Pilot 1-specific Activities
Major Sponsor Fast Track Activities
Pre-submission |
Post-submission |
|---|---|
Grant Fast-Track Designation Request |
Filing Meeting |
EOP1 Meeting |
Discipline Reviews |
EOP2 Meeting |
Mid-cycle review Meeting |
SPA |
Advisory Committee Meeting (if applicable) |
Pre-NDA/BLA Meeting |
Discipline Review Complete/Leadership Signoff |
|
Labeling (if applicacble) |
|
Action Letter |
Additional Sponsor Pilot-Specific Activities
Pilot Activities |
Recurring Activities |
"Startup" Activities |
|---|---|---|
Additional Filing meeting |
Some divisions have filing meeting for each RU |
Additional meetings to clarify Pilot process for the review team |
Pilot Admittance/RU Schedule neg. |
Additional meetings to determine eligibility of applications and to negotiate RU schedule |
Establishing which meetings (e.g., Filing) were necessary by RU |
Ack/Discipline Letters |
Sending an acknowledgement and discipline review letter for each RU |
Pilot acceptance decision-making process |
Internal FDA meetings |
Typical internal meetings may happen for each RU instead of only once in standard review |
Generating letter templates |
RU Re-work (RU were typically only reviewed once per cycle except amendments) |
Revisiting a discipline review in case of long lags between submissions (1 instance for 11 applications) |
|
While the incremental Pilot-specific activities did not occur for every Pilot product, in order to estimate the potential incremental effort for FDA, the analysis assumed a scenario in which the all the incremental activities would take place (i.e., worst-case/conservative scenario). To quantify the incremental effort, FDA data sources such as interviews, time tracking data, and the document storage system were used to capture those involved in the various Pilot activities. For each activity, the effort for discipline reviewers, regulatory project managers, team leaders, Division directors/deputy, office directors/deputy was estimated based on, for example, meeting preparation time, actual meeting time, and post-meeting efforts such as writing/reviewing meeting minutes for each review team member.
Based on this analysis, the total incremental effort for the Pilot was estimated to be between 190-360 direct labor hours per application. This effort is above what would typically be expended for a non-Pilot, Fast-Track/Rolling reviewed product in those Divisions included in the evaluation (i.e., experienced the Pilot 1 process). The breakdown by Pilot-specific activity is presented in Exhibit 4–16. The effort is displayed as a range, where the lower bars on the chart represent the low end range and the upper bars the high end range of effort as reported by review team members involved in the various activities.
Exhibit 4–16. Incremental Effort for FDA of Pilot-Specific Activities per Application (1 cycle)
Baseline effort= Non-pilot Fast-Track/rolling submissions within divisions with pilot products
| FDA Activities | Additional Filing Meetings | Pilot Application/ Negotiation | Acknowledgement/ DR Letters | Additional Internal Meetings | Start Up Effort | RU Re-work | Total Range |
|---|---|---|---|---|---|---|---|
| Lower Limit Effort | 44hrs | 42hrs | 35hrs | 24hrs | 33hrs | 12hrs | 190hrs |
| Upper limit Effort | 90hrs | 59hrs | 72hrs | 58hrs | 69hrs | 0 | 360hrs |
The effort is not distributed evenly among the review team members (Exhibit 4–17). Most of the incremental effort impacts the Regulatory Project Managers (RPMs), in charge of coordinating key review activities including: sponsor communications, scheduling reviewers, FDA-sponsor meetings, internal product-specific meetings and document reviews, and tracking PDUFA deadlines.
Exhibit 4–17. Incremental Effort of Pilot-Specific Activities by Function (1 cycle)
Baseline effort= Non-pilot Fast-Track/rolling submissions within divisions with pilot products
| FDA Roles | RPM | Reviewers | Team Leaders | Division Director/Deputy | Office Director/Deputy | Total Range |
|---|---|---|---|---|---|---|
| Lower Limit Effort | 53hrs | 66hrs | 45hrs | 23hrs | 3hrs | 190hrs |
| Upper limit Effort | 104hrs | 158hrs | 72hrs | 0 | 0 | 360 |
For the Divisions that participated in Pilot 1, Exhibit 4–18 shows the incremental effort for these Divisions to perform a Pilot 1 style review on all of their Fast-Track product applications. The estimated incremental effort increase is between 1,520-2,880 direct labor hours (based on the average number of Fast-Track applications submitted to those Divisions between FY 2003-2005).
Exhibit 4–18. Extrapolation of the Incremental Pilot Effort to the Average Number of Fast-Track Products Submitted to Divisions with Pilot 1 Experience
The estimated incremental Pilot 1 effort may not be evenly distributed among the Divisions with Pilot experience based on historical Fast-Track submissions. For example, between FY2003-FY2005, the Anti-infective and Ophthalmology and Drug Oncology Divisions received approximately 56% of all Fast-Track applications submitted in those fiscal years (Exhibit 4–19). Based on interviews with reviewers in those Divisions, to accommodate for this influx of Fast-Track applications, they strive to perform a six month review on all products considered a high priority, irrespective of the review designation. Because many of the procedures for early feedback and accelerated review have already been integrated, these Divisions may incur less Pilot 1 implementation effort (in the lower part of the range; Exhibit 3-18) compared to other Divisions. The estimated incremental increase may not be transferable to Divisions that did not have a Pilot 1 product or who do not have significant experience reviewing Fast Track applications. To understand the broader potential impact of expanding Pilot 1, FDA may consider prospectively monitoring the Pilot 2 products still in drug development and give sponsors the option to submit their applications as Pilot 1 products (if an application is submitted for those products). This would allow for further evaluation of the Pilot 1 effort, including the evaluation of 2 additional Divisions that were not included in this Pilot 1 evaluation.
Exhibit 4–19. Three Year Total Fast-Track Submissions by Divisions (FY2003-2005)
There is a possibility that the number of Fast-Track requests will increase if sponsors value the commitment of early review. This would obviously increase the estimated incremental effort shown in Exhibit 4–18.
If this increase were to occur, a major FDA concern is that important work without a PDUFA goal deadline may be shifted to a later date. Consequently, delaying such activities may have a negative impact on public health issues and other FDA responsibilities.
On balance, this workload prioritization challenge is a broader concern for the FDA, one that exists regardless of this Pilot program and especially with product reviews with a 6-month clock. The interviewed FDA Pilot participants did not report challenges completing other critical work specifically because of the Pilot. To caveat this finding, interviewees were primarily asked to compare the Pilot effort to their experiences with other Fast-Track, priority reviewed products and were not specifically asked about the shifting of non-PDUFA work. This is not to say, however, that high workload demands were not a concern, they were just not directly attributed to the implementation of the Pilot.
While this evaluation cannot validate or rule out the impact of non-PDUFA work being displaced, it is likely a broader concern for all products receiving a priority review. Therefore, additional evaluation may be warranted to determine the impact of the displacement of non-PDUFA work for products receiving a priority review.
Overall, this evaluation of the CMA Pilot 1 program does not confirm nor does it discard the possible positive impact on first-cycle approvals and reducing time to approval for important drug/biologic products. The FDA also incurred some additional effort from implementing and administering the Pilot program; however, it is recognized by the FDA that earlier communication of issues has potential value (this was also concluded in the Retrospective study cited earlier).
Specifically, FDA and sponsor Pilot 1 interviews as well as an earlier retrospective product application analysis, show that CMC issues have the greatest potential to be resolved quickly. Given earlier notification, the Sponsor would have a better chance of resolving such issues within the first cycle. Further, earlier detection and communication of issues is generally a prudent practice that may, over time, yield shorter times to approval.
From an FDA perspective, Pilot 1 did not offer robust positives; however, FDA reviewers involved in the Pilot program mentioned that the program was not overly burdensome and that the discipline reviews were effectively the same in terms of experience and effort (as compared to the Fast Track Program). The most significant FDA concerns are related to the increase in RPM workload, and the potential for non-PDUFA work (e.g., review of annual reports, new protocol reviews) being de-prioritized as a result of the review. In response to this general concern, reviewers who participated in the Pilot program did not explicitly express difficulty in fulfilling their responsibilities because of the Pilot. Further, the effects of re-prioritizing work for early submitted RUs may not be dramatic, as the FDA would have received portions of the application at a maximum, 12 months, but typically only less than 4-6 months earlier.
While largely not observed as part of this analysis, there is a possibility of additional work for reviewers within the first cycle Pilot 1 review as a result of conducting a fragmented review (blue circle, Exhibit 4–20) or if Sponsors rapidly address deficiencies identified early on, and submit amendments to resolve issues within the first review cycle (red circle, Exhibit 4–20). For the first scenario, only one reviewer of a Pilot product reported the need for such a re-review. This finding is likely the exception as the RU was received 13 months prior to complete submission which from both the FDA and Sponsors' perspective is rarely feasible. If under the second scenario early feedback ultimately leads to a first-cycle approval, then the increased first-cycle effort, may be off-set by not incurring the effort/costs of subsequent cycles (red circle, Exhibit 4–20).
Even if the need for a second review cycle is inevitable, a Pilot 1 submission may still reduce time to approval by offering more time to address deficiencies identified in early RUs. For the Sponsor, preparing the resubmission may be expedited while the FDA can potentially conduct a more focused second cycle review with a lower staff burden.
Exhibit 4–20. Scenarios on Potential FDA Re-Work
Sponsors mostly value the FDA commitment to provide early review/feedback. This not only offers additional time to address issues but also improves workload distribution and resource management. This benefit was of particular value to resource-constrained sponsors. Drawbacks included the uncertainty of product development preventing the sponsor from submitting timely RUs and the difficulty of preparing a comprehensive RU too far in advance.
In summary, due to several factors (e.g., the small sample size of the Pilot and the comparison cohort and the high unmet medical need nature of many of the products), there is no conclusive finding that indicates whether the Pilot 1 program should continue or be terminated. The key findings of the evaluation showed:
- Sponsors valued FDA's 6-month RU review commitment
- Helped distribute sponsors' workload
- Additional time to address issues for early submitted RUs is a review process benefit
- A strong first-cycle approval rate
- Similar level of application quality
- Similar levels of communication.
Given that this evaluation focused on the comparison of the Pilot 1 program to the Fast Track/Rolling Review program, many Pilot participants offered their perceptions of the Fast Track program. Industry strongly valued the subtle differences that Pilot 1 offered over the Fast Track/Rolling Review program where FDA remained more neutral.
While there is no resounding reason to continue the Pilot as a separate program, there may be merit to integrating some positive attributes/lessons-learned from the Pilot 1 program into the existing Fast-Track/Rolling Review structure. For example, some challenges with the current Fast Track/Rolling Review program are:
For these particular challenges, the Pilot 1 structure offers potential improvements over the Fast Track/Rolling Review program that include:
If the Fast Track/Rolling Review program were modified with these Pilot 1 attributes, this would allow the FDA to plan better for reviews because they can expect a complete section for early review; early review would be conducted consistently across FDA divisions for early submissions; and issues would be identified earlier, and in some cases, may lead to resolution prior to the first action date, or may help reduce the time between cycles if sponsors can begin addressing deficiencies earlier.
Some considerations before deciding to make any modifications may include:
Additionally, if modifications are implemented, other considerations may include:
Further, if the FDA decides to implement any modifications, additional resources would be required since the Agency would incur most of the additional workload burden. If implemented, the FDA may incur, in addition to the incremental costs described in this report, additional costs during the transition phase as this program is rolled out more broadly to the Divisions which in parallel need to complete reviews of applications currently under review. It is imperative that the FDA receive additional resources commensurate with the effort incurred to transition to and maintain the new process, in order to ensure that review Divisions are not overburdened.
Building on the interaction between the FDA and Sponsor during product development, the CMA Pilot 2 program promotes early scientific exchange in a more structured manner by formalizing certain interactions between the FDA and sponsors beginning with completion of Phase I through NDA/BLA submission.
At the time of evaluation, only one of nine drugs in the Pilot had submitted an NDA application (marking the end of the Pilot 2 process), thereby preventing a meaningful evaluation of the costs and benefits of CMA Pilot 2 to the drug development process. The other eight products are still in early to mid-stages of development, with many products having only just completed the first task of developing an Agreement. To the extent possible based on available data from both FDA sources and sponsors, the scope of this preliminary evaluation focused on:
Although it is too early to draw unequivocal conclusions, this evaluation highlights trends and significant observations that may provide guidance on the program effort and possible improvements to the Pilot 2 program. In order to fully assess the program's benefits and costs, continued monitoring and evaluation is necessary as products complete development and regulatory review.
This section addresses the Pilot 2 observations of FDA and sponsors, and is organized by the following subsections:
FDA and Sponsor interviews revealed that the first Pilot 2 activity-developing the meeting schedule agreement-was approached differently, which greatly influenced the interaction process and program effort. Two distinctive approaches were developed for sponsor/FDA interaction, the trigger method and the fixed schedule approach. Exhibit 5–1 details the two agreement approaches and FDA/Sponsor effort:
Trigger Method
Meeting schedules are linked
to the completion of milestones, and scheduling is ongoing as development
hurdles are achieved. Sponsors especially valued the certainty of receiving FDA
feedback during the drug development program with built-in schedule flexibility
while the FDA appreciated the program efficiency.
Fixed Schedule Method
This approach was the most
common interpretation and was also perceived to require relatively more effort
to execute. Under this approach, the FDA and Sponsor plan out all future
meetings/interactions with expected study completion dates. Participants using
the Fixed Schedule Method found this process to be onerous in light of received
benefits. In particular, the FDA RPMs and reviewers involved in this method
reported process concerns and unanimously agreed that this approach imposed a
great burden without adding much value. Specific difficulties mentioned were:
Exhibit 5–1. Details of the Meeting Schedule Agreement - Fixed vs. Trigger
|
Fixed Schedule Method |
"Trigger method" |
|---|---|---|
Description |
• Sponsor and FDA agree on schedule of meetings ahead of time • FDA is obligated to grant every meeting in the schedule • 7 of the 9 products followed this approach |
• Meetings can be requested with shorter timelines than currently the norm • Meetings are "triggered" by specific events in drug development process • FDA not obligated to grant every meeting requested |
Preparation Time |
• Multiple meetings required to negotiate schedule; negotiations take > 20 RPM and 20 reviewer hours • Must be renegotiated for each sponsor |
• Set of guidelines requires less preparation time than full schedule • Templates can be re-used for other products, eliminating the need to constantly reinvent |
Schedule Adherence |
• Meetings frequently rescheduled due to unpredictability in development • High cost for adjusting schedules when deadlines lapse |
• Flexible meeting schedule allows for meaningful discussion • Driven by specific drug development events |
Unnecessary Meetings |
• All planned meetings occur whether necessary or not |
• Meetings are triggered only if events occur • FDA retains some level of discretion in granting meetings • Avoids costly meetings |
In contrast, feedback from FDA teams involved in the Trigger Method suggested little additional effort was necessary to launch the program, and ongoing activities are not considered significantly different to how these Divisions typically interact with sponsors:
There are typically 3 types of FDA-Sponsor interactions that can occur during product development: Meetings in-person, teleconferences, and electronic communications (faxes & e-mails). To quantify program effort, the frequency of each interaction was determined from the Agreement and interactions documented in FDA's Document Filing System. For each type of interaction, an assessment of the average number of attendees by role was determined through FDA interviews. Next, an average level of effort (in hours) required to fully prepare, participate and follow through with any post-meeting action item was established for each person and type of interaction. Comparing corresponding data from the comparison cohort (non-pilot Fast-Track products received in 2003-2004), an overall program effort could be determined in hours by type of activity or by role.
Exhibit 5–2a shows the distribution of communication for the comparison cohort and Pilot 2 products. Dotted bars represent total communication counts based on the actual number that occurred to date, extrapolated to the full development cycle. Overall, FDA-Sponsor interactions have the potential to double compared to the routine interaction during the development stage of typical Fast-Track products (comparison cohort). Most significant are the additional in-person meetings introduced into the Pilot 2 program which represents the bulk of the newly incurred FDA effort (73%) - an estimated 376 hours out of the 512 additional hours required per Pilot 2 product (Exhibit 5–2b).
Exhibit 5–2. Analysis of the Fixed Schedule Method - Communication Increase and FDA Time Effort
Communication Type Frequency Actual/Planned) |
Meetings (In person) |
Telecon |
Electronic Communication (fax, letter, email) |
|---|---|---|---|
Pilot 2 |
9 |
5(10*) |
13 (26*) |
Fast-Track Comparison |
5 |
2 |
15 |
* Since development is underway for nearly all Pilot 2 products, the expected
number
of telecons
and electronic communications were extrapolated based on
number of
actual occurred through completion of the analysis.
|
Startup |
Meetings (In person) |
Telecon |
Electric Communications |
Total |
|---|---|---|---|---|---|
Estimated # hours for the Pilot 2 over the Baseline for Fast-Track |
77hrs |
376hrs |
36hrs |
23hrs
|
512hrs |
Sponsors ascribe high value to the commitment from the FDA for frequent communication and early feedback (Exhibit 5–3a). This input can provide a level of certainty in decision-making that may translate to easier planning and significant development cost and time savings (Exhibit 5–3b). Whether this input will translate into higher quality product development and ultimately better applications will have to be assessed upon completion of the development programs.
Exhibit 5–3. Sponsor Motivations for Program Participation and Perceived Benefits
As many products are still ongoing in the Pilot 2 program, evaluation topics such as meeting effectiveness and impact on PDUFA goals have yet to be determined. However, data has been collected on program implementation/maintenance effort and sponsor motivations. Both FDA reviewers and sponsors report positive attributes but also negative attributes associated with the program.
While most FDA Pilot participants agreed that the Pilot program could potentially influence the quality of applications from resource-constrained Sponsors, discipline reviewers are skeptical whether these will outweigh the negatives. The most common drawbacks mentioned included:
However, some Divisions reported that their typical operating procedure for any product, regardless of formal designation (e.g., Pilot 2, Fast-Track, non-Fast-Track), already closely mirrors the Pilot 2 process. Hence, these Divisions do not anticipate a dramatic increase in workload, if any.
Resoundingly, sponsors who participated in the Pilot program most value the commitment for timely FDA feedback and further attribute this benefit to their ability to better plan their product development program. This may result in avoiding costly delays and streamlining the product development process. In addition, these Sponsors reported that the cost in terms of effort and time of the Pilot program was minimal. Overall, sponsors valued the positive attributes greater than negative aspects.
A number of eligible sponsors however, chose not to apply for Pilot inclusion based on the perception that the level of FDA involvement during the IND would not significantly differ for the products under consideration (life-threatening diseases). These sponsors view the relevant FDA Divisions as sufficiently engaged and motivated. Of interest for these sponsors however, would be to enroll products that generally receive a "standard" review designation. It should be noted, that sponsors falling into this category were generally the multi-national pharmaceutical companies, with significant prior FDA experience across in many therapeutic areas.
Overall, early observations indicate two key factors are influencing Pilot 2:
Exhibit 4.4 summarizes some of these early observations that should be considered for continued monitoring and in case new INDs are accepted into the program.
Exhibit 5–4. Early Observations on the Pilot 2 Program
Scheduling |
• Provide guidance to sponsors on using the trigger method • Avoid delays and costs due to long negotiations of meeting schedule • Agreements should focus on the timing of FDA feedback for certain meetings/reviews |
|---|---|
Communication |
• The variability of communications across FDA divisions is a concern with all sponsors • Guidance on meetings (to be finalized in early '06) should help clarifiy "dos and don'ts" • Communicate problems upfront • Usage of "informal" communication can help clarify minor issues more quickly • Non-binding/open dialogue can help sponsors move in the right direction; in particular for study design |
Pilot Scope |
• Prospectively monitor the workload impact vs. value of the program |
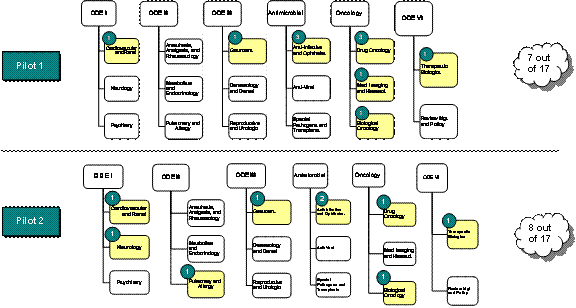
APPENDIX A: Distribution of Pilot 1 and Pilot 2 Products In FDA's Office of New Drugs
[1] Independent Evaluation of FDA's First Cycle Review Performance - Retroactive Analysis Final Report, December 14, 2005
![]()