|
 |
 |
|
 | |||||
| |||||||||
|
|
|
|
|
CDER Report to the Nation: 2005
Print version
3 International ActivitiesIndex
President’s Emergency Plan for AIDS ReliefTo meet our responsibilities to our own citizens we must increasingly look, think and act globally. We participate in harmonization committees. We are involved in bilateral and multilateral efforts to leverage scientific and financial resources with other nations to avoid duplication of effort and to cooperate in focusing on high-risk areas. President’s Emergency Plan for AIDS ReliefThe president’s $15 billion plan for AIDS relief around the world has a special focus on 15 countries hardest hit by the HIV epidemic. It targets three specific areas related to HIV/AIDS:
We are encouraging manufacturers to submit applications for fixed-dose combination and co-packaged versions of previously approved antiretroviral therapies. Tentative approval—whether for a new drug application or a generic drug application—will be the regulatory mechanism by which low-cost versions of innovator drugs sold in the developed world will become eligible for purchase under the emergency plan. Our tentative approval means that a drug meets our standards for safety, efficacy and quality but that existing patents or exclusivity prevent them from being sold in the United States. We have an expedited review process to ensure that the United States could provide safe, effective and affordable quality drugs to developing countries. We encouraged U.S. and foreign firms who were developing generic drugs to treat HIV disease to apply under the president’s plan . To meet plan's approval timelines, our generic drug reviewers implemented many process changes, including a rolling review approach. Our average review time for these applications has been six months. We lack information about most clinical laboratories and manufacturing sites associated with the products seeking approval under the emergency plan. Therefore, we also are engaged in outreach activities, manufacturer assistance, inspections and postmarketing monitoring. President’s plan focus countries
3-drug regimen gets tentative NDA approval for purchase under president’s planIn January 2005, within two weeks of receiving a complete application, we tentatively approved a complete three-drug product. It consisted of co-packaged lamivudine/zidovudine fixed-dose combination tablets and nevirapine tablets. We also approved a generic version of this regimen. Generic drugs eligible for purchase under president’s planAs of June 29, 2006 we had fully approved two generic drugs and tentatively approved another 19. A list and more information is at http://www.fda.gov/oia/pepfar.htm. Information-Sharing AgreementsBecause of enhanced cooperation among regulators around the world, FDA has entered into international agreements in which we play a critical implementation role. We have a growing list (left) of regulatory partners worldwide with whom we can pursue more open dialogue on emerging issues as well as exchange routine information on scientific review, policy development and enforcement. Examples or our agreements include: Japan and AustraliaWe routinely exchange recall information about products of interest to Japan and Australia and communicate emerging enforcement activities of mutual interest. We continue to collaborate with our counterparts regarding site inspection information. With limited inspection resources of our own, we increasingly depend on foreign regulatory inspections and incorporate their inspection findings into a risk-based program for future inspection. European Medicines Agency (EMEA)This agreement establishes a basis for exchanging confidential information with the European agency primarily responsible for approving drugs. It permits our review and compliance staff to share important information about pending approvals, post-marketing surveillance and enforcement actions concerning products and facilities under the European agency’s oversight. Implementation, to be phased in, includes activities to build understanding and mutual confidence in one another’s systems. Mexico and CanadaFDA is working jointly with our North American neighbors to develop information exchange arrangements about drug manufacturing facilities in each of our countries and to share information about product recalls that may impact our consumers. Our recent contributions to this long-standing effort have been vital in moving this relationship in a mutually beneficial direction. Exchanges of product recalls, emerging compliance issues and site-specific information have already begun. An agreement with Canada provides for the exchange of information about pending approvals, post-marketing surveillance and enforcement actions. SwitzerlandThe working arrangement with Switzerland began several years ago and has continued to progress steadily in 2005. The present agreement addresses the need for protection of confidential information and provides for the exchange of information about marketing approval decisions, post-market surveillance, policy developments and compliance or enforcement activities of mutual interest. Progress in implementing this arrangement includes the exchange of technical staff and training opportunities as well as inspection information. Successful joint inspections have helped foster mutual confidence and improve communications. International agreementsn Countries
n Organizations
International regulators forumIn 2005, we held the first of a series of twice yearly week-long forums with international regulators. There were 27 representatives from all but one of the 15 PEPFAR focus countries, 19 from seven other African and Asian countries and three from the World Health Organization. We provided information about U.S. drug regulatory processes and shared perspectives on approaches to common regulatory and scientific challenges. Pharmaceutical Inspection Cooperation SchemeAs part or of our initiative to improve manufacturing practices, FDA applied for membership in this international organization dedicated to drug regulatory harmonization and collaboration in the area of good manufacturing practices. CGMP workshop in ChinaTo foster compliance with current good manufacturing practices, we co-sponsored an educational public workshop in collaboration with Peking University and the International Society for Pharmaceutical Engineering. Assuring International Trade QualityWhile the globalization of pharmaceutical commerce brings the benefits of modern drugs to citizens worldwide, it poses many challenges to us and regulators in foreign countries. We share with them a common interest in ensuring our citizens have access to safe, effective and high quality products and are protected from counterfeit drugs and terrorist threats. Drug exportsExport certificates attest that U.S. drug products are subject to inspection by FDA and are manufactured in compliance with current good manufacturing practices. These certificates enable American manufacturers to export their products to foreign customers and foreign governments. The demand for certificates remains high due to expanding world trade, ongoing international harmonization initiatives and international development agreements. Export certificatesWe issue export certificates that verify the drug products being exported: n Were freely marketed in the United States. n Were in compliance with U.S. laws and regulations. n Met certain national or international standards, such as quality standards. n Were free of specific contaminants.
Click image for larger chart, click here for accessible text. Drug importsAgency resources are particularly focused on counterfeit drugs and counterterrorist activities. We work to: n Enforce the law. With FDA’s field force, we enforce legal requirements determining which drug products may be imported by manufacturers, distributors and consumers. n Identify and interdict illegal drugs. We take steps to ensure that imported drugs are not counterfeit, unapproved, adulterated or misbranded and that they meet applicable legal requirements relating to safety and effectiveness. n Improve technology. Along with the pharmaceutical and advanced technology industries, the states and other federal agencies, we are monitoring the development and implementation of “track and trace” technology that will enhance anti-counterfeiting measures by providing real-time monitoring of a drug product through the U.S. drug distribution system. Foreign inspectionsn 234 preapproval inspections in support of:
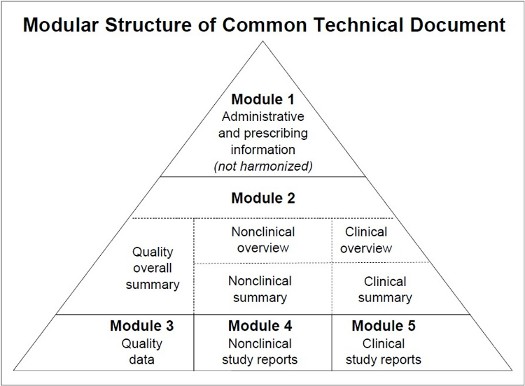
n 213 current good manufacturing practice inspections For most foreign inspections, both a CGMP and a preapproval inspection take place and are counted twice, once under each inspection program. Our review of 263 inspection requests for foreign establishments, resulted in three warning letters, two import alerts and several regulatory meetings. HarmonizationHarmonization—making the drug regulatory processes more efficient and uniform—is an issue that is important not only to Americans, but to drug regulatory agencies and pharmaceutical companies throughout the world. The International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use has worked to bring together government regulators and drug industry experts from innovator trade associations in the European Union, Japan and the United States. We are leading the FDA's collaboration with the ICH. This work is making new drugs available with minimum delays not only to American consumers but also to patients in other parts of the world. The drug regulatory systems in all three regions share the same fundamental concerns for the safety, efficacy and quality of drug products. Before ICH, many time-consuming and expensive technical tests had to be repeated in all three regions. The ICH goal is to minimize unnecessary duplicate testing during the research and development of new drugs. The ICH process results in guidance documents that create consistency in the requirements for product registration in the three regions. Common Technical DocumentThe ICH Common Technical Document allows data in the same format to be submitted to drug review authorities in all three ICH regions. Specifications for electronic submission of the CTD, known as the eCTD, were completed in 2002.
Click here for accessible description. Internet resourceMore information is on the ICH Web site at http://www.ich.org. Electronic Common Technical DocumentElectronic submissions using the eCTD can be used to submit all applications and related submissions such as promotional materials and adverse events. Among other things, the eCTD allows reviewers to: n Create an up-to-date, cumulative table of contents for the entire application at any time. n Access any electronic submission from a single screen. n Download files so submissions can be used even when the reviewer is off the network. Harmonization guidancesWe publish International Conference on Harmonization documents as guidances to industry on our Web site at http://www.fda.gov/cder/guidance/index.htm. As of June 12, 2006, we had: n 55 final guidances
n 5 draft guidances
Internet resourcesMore information about our international activities, including Spanish language materials, is at http://www.fda.gov/cder/audiences/iact/iachome.htm.
Date created: August 18, 2006 |