|
 |
 |
|
 | |||||
| |||||||||
|
|
|
|
|
CDER Report to the Nation: 2005
Print version
Drug Review (continued)Index
Pediatric Drug DevelopmentThe Best Pharmaceuticals for Children Act of 2002 (BPCA) renewed our authority to grant six months of additional marketing exclusivity to manufacturers who conduct and submit pediatric studies in response to our written requests. In calendar year 2005, we approved 25 pediatric labeling changes as a result of studies conducted under the exclusivity provision. Pediatric exclusivity. As of April 30, 2006, we had received 467 proposed pediatric study requests from manufacturers, issued 320 written requests, made 130 exclusivity determinations, granted exclusivity to 118 drugs and added new pediatric information to 109 labels. Improved safety, dosing information. About one-fourth of the new pediatric labels have safety or dosing information. We are discovering important differences between adults and children in the clearance and metabolism of drugs. Underdosing leads to ineffective treatment, and overdosing poses a greater risk of adverse reactions. Pediatric safety signals identified in these studies include effects on growth, school behavior, suppression of the adrenal gland and suicidal ideation. The failure to produce drugs in dosage forms that can be taken by young children—such as liquids or chewable tablets—can deny them access to important medications. As a result of pediatric testing we now have 12 drugs with new pediatric formulations and six drugs with recipes in their labels to provide directions for the pharmacist to compound an age-appropriate formulation. Off-patent drugs. The law also established a publicly funded contracting process to study drugs that lack patent protection or market exclusivity, referred to as “off-patent.” In consultation with FDA and other pediatric experts, the National Institutes of Health have published five lists of off-patent drugs for which additional pediatric studies are needed. To date, we have issued 17 written requests—five in 2005—for these off-patent drugs. We also have forwarded eight written requests – two in 2005-for on-patent drugs, for which sponsors declined pediatric studies. Public disclosure. We publish a summary of the medical and clinical pharmacology reviews of the pediatric studies conducted under the law. As of April 30, 2006, we have posted 60 summaries, regardless of the regulatory action, at http://www.fda.gov/cder/pediatric/Summaryreview.htm. Adverse events reported. The act mandates review of all adult and pediatric adverse event reports for a one-year period after pediatric exclusivity is granted. The reviews of the reports are then presented to the Pediatric Advisory Committee. As of April 2006, reports for 54 drugs have been presented. Significant pediatric safety signals have been found, including neonatal withdrawal with antidepressant use during pregnancy and serious adverse events, including deaths, due to fentanyl transdermal use in children. Pediatric Research Equity Act of 2003In September 2005, we published a draft guidance on how to comply with the law. The law gave us the authority to require pediatric studies of certain new drugs and biological products when such studies are needed to ensure the safe and effective use of the products in children. However, the law does not require the same public disclosure of pediatric studies required under the Best Pharmaceuticals for Children Act. 2005 new approvals with pediatric indicationsn New molecular entities. Two orphan treatments for children who are very short for their age and an iron chelating agent indicated for patients 2 and older. n Biologics. An enzyme replacement therapy for a rare inherited condition that can affect children. n New drugs. An oral solution of an antiretroviral medicine and four over-the-counter drugs. Safety issues for children’s usesWe issued Public Health Advisories about pediatric safety issues including: n A boxed warning about using the eczema therapies pimecrolimus and tacrolimus only as directed and not at all in children younger than 2 years of age. n Suicidal thinking in children and adolescents treated with atomoxetine for attention deficit hyperactivity disorder. We added a black box warning about suicidality in children and adolescents to the labeling for antidepressants. Full information and a list of the drugs is at http://www.fda.gov/cder/drug/antidepressants/default.htm. 2005 pediatric drug statistics
Click image for larger chart, click here for accessible text. Notable 2005 pediatric new or expanded usesThe Best Pharmaceuticals for Children Act requires us to give priority status to pediatric supplements for drug studies submitted in response to a written request. An efficacy supplement may change labeling to reflect new information about pediatric use, even if there are no new or expanded uses. The review of these pediatric supplements resulted in labeling that describes new and expanded uses of these medications in children: Ertapenem sodium (Invanz), an injectable antibiotic, had its labeling expanded to include the treatment of susceptible moderate to severe infections in children 3 months of age and older. Clinical pharmacokinetic studies proved that ertapenem is not recommended in the treatment of meningitis in the pediatric population due to lack of sufficient CSF penetration. Ethinyl estradiol and norgestimate (Ortho Tri-Cyclen), an oral hormone combination, had its labeling revised to indicate that there was no significant difference between treatment and placebo in bone mineral density in 123 adolescent females with anorexia nervosa in a one-year clinical trial. Insulin aspart (NovoLog), a rapid-acting injectable human insulin analog, labeling was expanded to include treatment of patients 6 to 18 years old with type 1 diabetes. Levetiracetam (Keppra), available as tablets and oral solution, labeling was expanded to include use as adjunctive therapy in the treatment of partial onset seizures in pediatric patients 4 years and older with epilepsy. Meloxicam (Mobic), a nonsteroidal anti-inflammatory drug available as tablets and oral suspension, labeling was expanded to include treatment of certain types of juvenile rheumatoid arthritis in patients 2 years of age and older. Mixed salts of a single entity amphetamine (Adderall XR), a once or twice daily extended-release central nervous system stimulant, labeling expanded to include treatment of attention deficit hyperactivity disorder in patients 13 to 17 years of age. Previously approved for use in children 6 to 12 years old. Ondansetron (Zofran), an antiemetic available as tablets and an injectable, labeling expanded to include dosing in children down to 6 months of age for the prevention of chemotherapy-induced nausea and vomiting, as well as dosing in children down to 1 month of age for the prevention of postoperative-induced nausea and vomiting. Labeling previously provided dosing down to 4 years of age for the prevention of chemotherapy-induced nausea and vomiting and dosing down to 2 years of age for the prevention of postoperative-induced nausea and vomiting. Oxcarbazepine (Trileptal), an anticonvulsant available as tablets and oral suspension, labeling expanded to include treatment as adjunctive therapy in children aged 2 years and above with epilepsy. Previously approved for children 4 years and older. In clinical studies, it was observed that children 2 to <4 years of age may require up to twice the dose per body weight compared to adults. Sibutramine (Meridia), a weight loss aid, labeling modified to reflect that data from a clinical study in obese adolescents are inadequate to recommend the use of sibutramine for the treatment of obesity in pediatric patients. In addition, important safety information was added. Notable non-BPCA pediatric new or expanded useNitazoxanide (Alinia) can be used to treat diarrhea caused by Cryptosporidium parvum in non-HIV infected patients 12 years of age and older. 2005 priority pediatric labeling changesOur priority review of these pediatric supplements was consistent with the BPCA unless noted:
Internet resourcesOur Web site for up-to-date pediatric labeling changes is at http://www.fda.gov/cder/pediatric/index.htm. Pregnancy and Lactation LabelingTo improve our knowledge of the use of drugs during pregnancy and lactation, we sponsor research and provide scientific guidance to industry and our reviewers. Women who are pregnant often need to use prescription medicines. In many cases, a disease or condition left untreated may be more harmful to a woman and her fetus or nursing baby than a drug treatment. In other cases, a different drug treatment than she is already on may be safer. We have reviewed the current system of labeling drugs for use by pregnant and lactating women and are developing an improved, more comprehensive and clinically meaningful approach. We are consulting with government agencies, medical experts, consumer groups and the pharmaceutical industry to develop this new labeling format. We work with our reviewers and pharmaceutical companies to update product labels with available human data regarding exposure to drugs during pregnancy and lactation. Scientific guidancen Risks of drug exposure in human pregnancies. In 2005, we issued our final guidance for our reviewers on how to evaluate human data on the effects of in utero drug exposure on the developing fetus. n Lactation studies in women. In 2005, we published a draft guidance for industry that provides the basic framework for designing, conducting and analyzing clinical lactation studies. n Determining the appropriate dose of a drug for pregnant women. In 2004, we published a draft guidance for industry that provides the basic framework for designing, conducting and analyzing pharmacokinetic and pharmacodynamic studies in pregnant women. n Pregnancy exposure registries. In 2002, we published a final guidance for industry that provides advice on how to establish registries that prospectively monitor the outcomes of pregnancies in women exposed to a specific drug. These registries can provide clinically relevant human data for treating or counseling patients who are pregnant or anticipating pregnancy. Scientific research in pregnancy and lactationWe funded several studies to evaluate either fetal safety from drug exposure or whether the dose of a drug should be adjusted during pregnancy or lactation: n Counter-terrorism. These studies look at specific anti-infective drug products that would be used for treatment following exposure to specific bioterrorism agents. They focus on use in special patient populations, such as women who are pregnant or lactating and the elderly. They evaluate either the need for dose adjustments in these special patient populations or fetal safety following in utero drug exposure. n Liver enzymes. These studies look at the effects of pregnancy on specific drug-metabolizing enzymes in the liver. Research on drugs for high blood pressure, depressionFDA’s Office of Women’s Health funded studies to look at specific drugs used to treat high blood pressure and depression and determine if the doses of these drugs should be adjusted during pregnancy. Over the Counter Drug ReviewOver-the-counter drug statistics
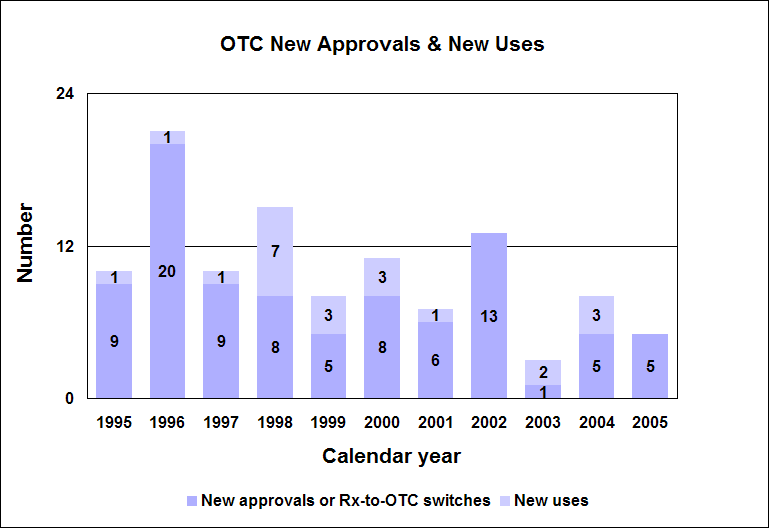
Click image for larger chart, click here for accessible text. Over-the-Counter Drug ReviewWe approved five new drug applications for first-time over-the-counter sale. There were no prescription-to-OTC switches or new uses approved in 2005. Our OTC approvals were: n Ibuprofen and diphenhydramine hydrochloride (Advil PM Liquigels and Caplets) for relief of occasional sleeplessness associated with pain in adults. n Loratadine (Loratadine Oral Suspension) for the temporary relief of these symptoms due to hay fever or other upper respiratory allergies: runny nose, sneezing, itchy, watery eyes, itching of the throat or nose in adults and children 2 years of age and older. n Chlorhexidine gluconate (2% Chlorhexidine Gluconate Cloth) for use as a patient preoperative skin preparation in adults and children 2 months of age and older. n Loperamide (Loperamide HCl soft gelatin capsules) to control symptoms of diarrhea, including traveler’s diarrhea in adults and children 6 years of age and older. n Chlorhexidine gluconate and isopropyl alcohol (Chlorascrub Swab, Chlorascrub Swabstick and Chlorascrub Maxi Swabstick) for use as a patient pre-injection preparation and as a patient preoperative skin preparation (Chlorascrub Swabstick and Chlorascrub Maxi Swabstick) in adults and children 2 months of age and older. Office of Nonprescription Products createdWe created an Office of Nonprescription Products in our reorganization of the Office of New Drugs. Within the office, the Division of Nonprescription Clinical Evaluation focuses mainly on the review of investigational new drugs and new drug applications. The Division of Nonprescription Regulation Development focuses mainly on the development of OTC drug monographs. Improved labels for OTC medicinesAmerican consumers are benefiting from easy-to-understand labels on drugs they buy without a prescription. A mandatory changeover to the new labels, titled “Drug Facts,” began in 2002 and is now complete for all products, with a few exceptions, as of May 2005. How we regulate OTC drugsWe publish monographs that establish acceptable ingredients, doses, formulations and consumer labeling for OTC drugs. Products that conform to a final monograph may be marketed without prior FDA clearance. Drugs also can be approved for OTC sale through the new drug review process. More information about the OTC drug review process is at http://www.fda.gov/cder/about/smallbiz/OTC.htm. Education campaign on safe use of OTCsWe developed a national education campaign to provide advice on the safe use of over-the-counter pain and fever reducers (http://www.fda.gov/cder/drug/analgesics/). Because many OTC medicines for different uses have the same active ingredients, an unintentional overdose is possible. We are focusing on OTC drug products that contain acetaminophen and non-steroidal anti-inflammatory agents, which include products such as aspirin, ibuprofen, naproxen sodium and ketoprofen. Generic Drug ReviewGeneric drug statistics
Click image for larger chart, click here for accessible text.
Click image for larger chart, click here for accessible text.
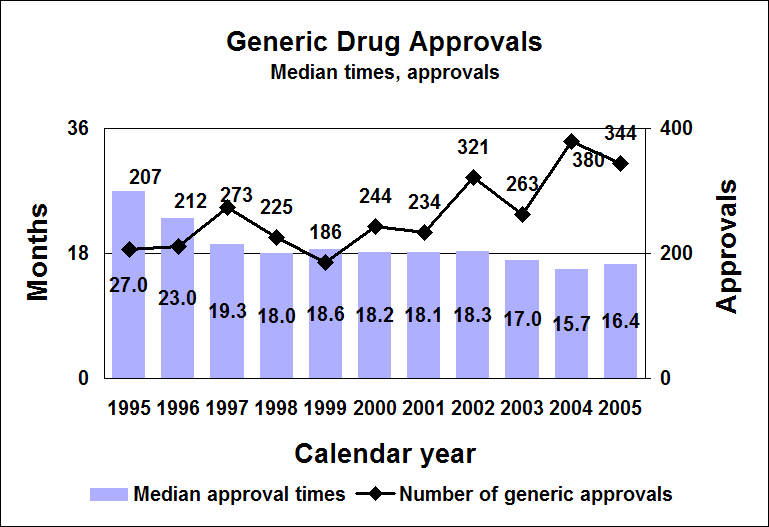
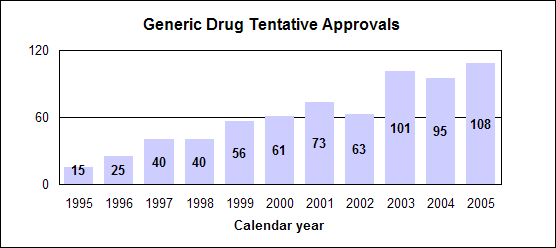
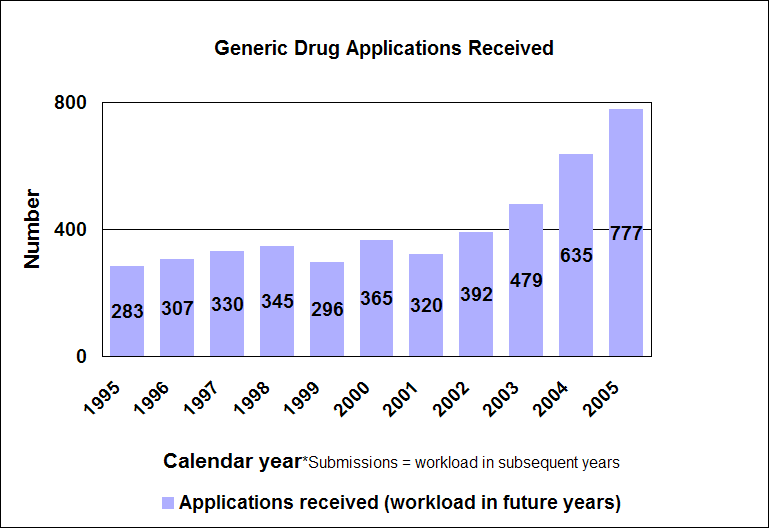
Click image for larger chart, click here for accessible text. We approved 344 generic drug products in 2005, including a substantial number of products that represent the first time a generic drug was available for the brand-name product. The median approval time was 16.4 months. The median statistic for total approval time had hovered at about 18 to 19 months for six years. We have made several changes to improve the efficiency of our generic drug review process in order to try to keep up with the dramatic increase in applications. These efforts will continue. Notable 2005 generic drug approvalsExamples of first-time generic drug approvals are: n Azithromycin. An antibiotic used for the treatment of mild to moderate infections of various types. n Fentanyl transdermal system. Used for managing chronic pain. n Fexofenadine. Used to relieve seasonal allergic rhinitis symptoms. n Levofloxacin. A broad spectrum antibiotic used for various conditions such as pneumonia, bronchitis and sinusitis. n Ramipril. Used to treat high blood pressure, heart failure after a heart attack and risk reduction for certain cardiac events. n Zidovudine. Used in combination with other antiretroviral agents for the treatment of HIV infection. Our approval of generic versions of these drugs can save American consumers and the federal government hundreds of millions of dollars each year.Tentative vs. full approvalThe only difference between a full approval and a tentative approval is that the final approval of these applications is delayed due to an existing patent or exclusivity on the innovator drug product. These and other legal issues continue to be a challenge to the generic drug review program. The review of an application that is tentatively approved requires the same amount of work as a review that results in a full approval. While tentative approvals represent a full workload for us, they are only displayed in our approvals chart once they are converted to full approvals. For example, some of the approvals in 2005 represent conversions of tentative approvals granted in 2004 or previous years. Tentative approvals key to affordable, worldwide AIDS reliefTentative approval is a key regulatory mechanism to support the availability of drugs for the President’s Emergency Plan for AIDS Relief. How we approve generic drugsGenerics are not required to repeat the extensive clinical trials used in the development of the original, brand-name drug. For many products such as tablets and capsules, the generics must show bioequivalence to the brand-name reference listed drug. This means that the generic version must deliver the same amount of active ingredient into a patient’s bloodstream and in the same time as the brand-name reference listed drug. The rate and extent of absorption is called bioavailability. The bioavailability of the generic drug is then compared to that of the brand-name. This comparison is bioequivalence. Brand-name drugs are subject to the same bioequivalency tests as generics when their manufacturers reformulate them. Scientific basis for generic drug reviewWe continue to articulate the scientific underpinnings of our review process and to work to define mechanisms to evaluate equivalence of certain unique products. Online educationWe are offering a free online educational tutorial on the generic drug approval process that offers one hour of continuing education credit for certain health professionals. The course, available at http://www.connectlive.com/events/genericdrugs/, educates health professionals on how our approval assures that generic drugs are safe, effective and high quality products. Improving manufacturing practicesThe strategic initiative to develop a 21st century drug quality system also applies to generic drugs. Consumer communicationOur efforts to build consumer confidence in generic drug products are continuing through our Generic Drug Quality Awareness program. We have partnered with a number of professional and consumer organizations to launch programs about the quality and benefits of generic drugs. We have helped design messages that appear on prescription bags in chain drug stores. Radio public service announcements with the generic drug quality message will be appearing in several geographic areas. Our generic drug public service announcements are at http://www.fda.gov/cder/consumerinfo/generic_info/default.htm. Generic drug Web siteYou can find more information about our generic drug program at http://www.fda.gov/cder/ogd/. Generic drug review efficienciesThe dramatic increase in receipts of generic drug applications makes it imperative that we process generic drug applications more efficiently. With the overall goal of getting generic drug products to the consumer as efficiently as possible, we continue to look for ways to improve our processes and also to provide communication and guidance to industry. We are taking steps aimed at improving the content and completeness of generic drug applications and assuring that the applications contain the needed information to be evaluated successfully in one cycle. These steps include: n Enhanced communication with individual applicants during the review process. n Working with the generic drug industry association to help their members submit applications that can be reviewed more efficiently. n Exploring further enhancements to the review process. n Holding joint meetings and workshops with industry to enhance knowledge of topics of interest. n Efforts to encourage submission of applications in an electronic format for greater efficiency. Electronic submissionsThrough public presentations, we are encouraging the generic drug industry to submit their applications electronically. Restructuring to meet increased demandsWe have constituted a third chemistry review division for generic drugs. We also are augmenting our clinical, microbiology and bioequivalence review staffs to meet the needs for review of a growing number of generic drug applications. Reducing hurdles to generic drug availabilityWe are working on regulations to decrease time-consuming legal delays in the approval and marketing of generic products. These rules, implementing provisions of the 2003 Medicare Prescription Drug, Improvement and Modernization Act, will: n Limit an innovator firm to one 30-month delay for courts to resolve patents challenged by an generic manufacturer. n Prevent a generic manufacturer with 180-day exclusivity from delaying marketing in order to deny other generic firms entry into the market. More generic competition results in lower drug pricesThe entry of a second generic competitor brings about the largest price reduction. We concluded this from our analysis of IMS retail sales data for single-ingredient brand-name and generic drug products sold from 1999 through 2004. Our study is at http://www.fda.gov/cder/ogd/generic_competition.htm.
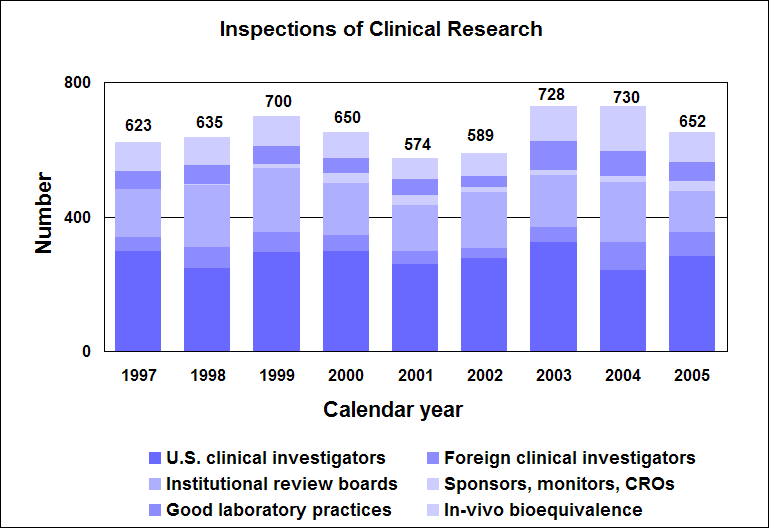
Click image for larger chart, click here for accessible text. Assessing Data Quality, Research RisksInspections for data quality, research risks in 2005We conducted a total of 652 inspections in 2005:
Click image for larger chart, click here for accessible text. When obtaining data about the safety and effectiveness of drugs, sponsors rely on high quality laboratory studies and human volunteers to take part in clinical studies. Protecting volunteers from research risks is a critical responsibility for us and all involved in clinical trials. We perform on-site inspections to protect the rights and welfare of volunteers and verify the quality and integrity of data submitted for our review. We inspect domestic and foreign clinical trial study sites; institutional review boards; sponsors, monitors and organizations conducting research; laboratories that obtain data; and sites performing bioequivalence studies in humans and preclinical studies in animals. Our programs to protect volunteers are challenged by increases in the number of clinical trials, the types and complexity of products undergoing testing, and the increased number of trials performed in countries with less experience and limited or no standards for conducting clinical research. Sponsors and clinical investigators protect volunteers by ensuring that: n Clinical trials are appropriately designed and conducted according to good clinical practices. n Research is reviewed and approved by an institutional review board. n Informed consent is obtained from participants. n Ongoing clinical trials are actively monitored. n Special attention is given to protecting vulnerable populations, such as children, the mentally impaired and prisoners. We require sponsors to disclose financial interests of clinical investigators who conduct studies for them. This helps identify potential sources of bias in the design, conduct, reporting and analysis of clinical studies. Top 5 deficiencies found during inspections of clinical investigators in 2005
International inspections of clinical researchWe conducted 70 inspections of clinical research in 25 countries in 2005. We participate in international efforts to strengthen protections for human volunteers worldwide and encourage clinical investigators to conduct studies according to the highest ethical principles. This includes our work with the International Conference on Harmonization and the Declaration of Helsinki. Internet resourcesMore information on data integrity and patient safety is at http://www.fda.gov/cder/offices/dsi/index.htm. Electronic SubmissionsWe cooperated with outside organizations working to publish standards for submitting study data. These groups include the Clinical Data Interchange Standards Consortium and Health Level 7, also known as CDISC and HL-7 respectively. Some of these projects are: n Clinical trial data. We adopted the consortium’s Study Data Tabulation Model version 1.0 for submission of information from clinical trials. n Preclinical data. The consortium is working to extend the model to handle animal toxicity and microbiology data. n Database development. We completed a database model for storing and accessing both clinical and animal toxicity data submitted using the Study Data Tabulation Model. We are collaborating with the National Cancer Institute and software vendors to implement the database and develop “smart” tools for accessing the data. n Electrocardiogram data. We adopted the Health Level 7 standard for annotated electrocardiogram waveform data. We are working with a vendor to develop software for analyzing the data and a warehouse for storing it. n Structured product labeling. We are accepting Health Level 7 Structured Product Labeling for content of labeling submissions. We are developing a repository for storing the data and software to improve the processing and reviewing of labeling changes. This is part of our effort to improve patient safety through access to the most recent information about medicines. We continue to receive electronic submissions using the specifications of the electronic Common Technical Document. Electronic submission of labeling requiredIn 2005, we issued final guidance to assist manufacturers in submitting prescription drug label information to us in a new electronic format. These electronic product labels are the key element and primary source of medication information for “DailyMed”—a new interagency online health information clearinghouse that will provide the most up-to-date medication information free to consumers, health-care providers and health-care information providers. Using embedded computer tags, the prescribing and product information can be electronically managed, allowing a user to search for specific information. These tags can instruct computers to read specific sections of a drug label including product names, indications, dosage and administration, warnings, description of drug product, active and inactive ingredients, and how the drug is supplied. With this information, physicians will be able to quickly search and access specific information they need before prescribing a treatment, resulting in fewer prescribing errors and better informed decision making. In addition, the electronic labels will improve our drug labeling review process, so that we can provide immediate access to the most recent information about medications to prescribers and patients.Internet resourcesMore information on our electronic submissions program is at http://www.fda.gov/cder/regulatory/ersr/. User Fee ProgramAmericans deserve timely access to potentially lifesaving new drugs as soon as possible once they are proven safe and effective. The Prescription Drug User Fee Act of 1992 received its second five-year extension in 2002, known as PDUFA III. This reauthorization is helping us ensure that we have the expert staff and resources to review applications promptly and get safe, effective new drugs into the hands of the people who need them. The current user fee law maintains our high review performance goals, includes increased consultations with drug sponsors and provides for earlier feedback on their submissions. User fee performanceUnder legislation authorizing us to collect user fees for drug reviews, we agreed to specific performance goals for the prompt review of submissions. n We exceeded all our performance goals for the fiscal year 2004 receipt cohort. n We are on track for exceeding most user-fee performance goals for the fiscal year 2005 cohort. Continuous marketing application pilot programsUnder PDUFA III, we are assessing the value of both early review of parts of marketing applications and of more extensive feedback to sponsors during their development programs. Two pilots for “continuous marketing applications” apply to drugs and biologics in our fast track program: n Pilot 1 allows applicants to submit predefined portions of their marketing applications called “reviewable units” before submitting the completed application. Each reviewable unit has a six-month goal for issuing a discipline review letter. In fiscal year 2005, we met our performance goal for reviewable unit submissions. n Pilot 2 allows us to enter into agreements with sponsors for frequent scientific feedback and interactions during the clinical trial phase of product development. As of Aug. 1, 2005, there were nine development projects entered in the Pilot 2 program. The pilots have limitations and specific criteria for entry. More information is available at http://www.fda.gov/cder/pdufa/CMA.htm. Internet resources for user feesOur user fee Web site at http://www.fda.gov/cder/pdufa/default.htm has links to PDUFA:
Drug Review TeamWe use project teams to perform reviews. Team members apply their individual special technical expertise to review applications: n Biologists, biochemists and immunologists evaluate the manufacturing processes for biological products to ensure the continued purity, potency and safety of these products. They also provide insights to the review team regarding the mechanism of action and potential and observed adverse events associated with specific products. n Chemists focus on how a drug is manufactured. They make sure the manufacturing controls, quality control testing and packaging are adequate to preserve the drug product’s identity, strength, potency, purity and stability. n Clinical pharmacologists and biopharmaceutists evaluate factors that influence the relationship between the body’s response and the drug dose and evaluate the rate and extent to which a drug’s active ingredient is made available to the body and the way it is distributed, metabolized and eliminated. They also assess the clinical significance of changes in the body’s response to drugs through the use of exposure-response relationships and check for interactions between drugs. n n Microbiologists evaluate the effects of anti-infective drugs on germs. These medicines—antibiotics, antivirals and antifungals—differ from others because they are intended to affect the germs instead of patients. Another group of microbiologists evaluates the manufacturing processes and tests for sterile products, such as those used intravenously. n Pharmacologists and toxicologists evaluate the effects of the drug on laboratory animals in short-term and long-term studies, including the potential based on animal studies for drugs to induce birth defects or cancer in humans. n Physicians evaluate the results of the clinical trials, including the drug’s adverse and therapeutic effects, and determine if the product’s benefits outweigh its known risks at the doses proposed. n Project managers orchestrate and coordinate the drug review team’s interactions, efforts and reviews. They also serve as the regulatory expert for the review team and as the primary contact for the drug industry. n Statisticians evaluate the designs and results for each important clinical study. Scientific training for reviewersOur systematic, internal training program is based on core competencies, learning pathways and individual development plans. In 2005: n We presented 30 scientific seminars and scientific rounds. n We offered a strong and innovative curriculum of 45 scientific courses. n We brought in 49 visiting professors to talk directly to individual review divisions about critical, new drug-related research and techniques. n We offered additional courses in job skills, research tools, leadership and management. Advanced scientific educationA committee of our scientists oversees a program of scientific training, seminars, case study rounds and guest lectures. This multidisciplinary program helps keep our scientists up-to-date on the latest developments in their fields and current industry practices. Academics to CDEREach spring, we collaborate with five local universities to present an up-to-date course on a compelling scientific topic. Recent topics were:
n
2005: Critical path science
Date created: August 18, 2006 |