


CBER Presentation
FDA Review of Investigational Cell-Based Therapies: Resource Tools, Framework and Issues
Donald W. Fink, Jr., Ph.D.
Phone (301) 827-5153
E-mail: donald.fink@fda.hhs.gov
Office of Cellular, Tissue and Gene Therapies
Division of Cellular and Gene Therapies
The Stem Cell Meeting
March 14, 2006
Challenges on the Horizon
Recent Examples from Scientific Literature
- Stem cells for cardiac repair: state of the art. Front Biosci. 01-September-2005, Vol 10, pg. 3143-3150-12746 (Condorelli and Peschle).
- Superior ex vivo cord expansion following co-culture with bone marrow-derived msesnchymal stem cells. Bone Marrow Transp. Feb, 37(4):359-366, 2006 (Shpall EJ et al.).
- Derivation of clinical-grade human embryonic stem cells. Reprod Biomed Online, Jan, 12(1):112-118, 2006 (Simon C et al.).
- Regulation of mesenchymal stem cell adhesion and orientation in 3D collagen scaffold by electrical stimulus. Bioelectrochem. 2006, Feb 9 (Cho et al., Epub ahead of print).
- Lentiviral vector-mediated transduction of neural progenitor cells before implantation into injured spinal cord and brain to detect their migration, deliver neurotrophic factors and repair tissue. Restor Neurol Neurosci., 23(5-6):313-324, 2005 (Bunge et al.)
Topics to be Covered
Paradigm: Novel Cellular Therapies Including Stem Cells
- Important Tools/Resources that Support the Review Process
- Issues Critical to the Regulation of Stem Cell Based Therapies
- Helpful Hints
- Selected Guidance Documents and Contact Information
Resources Important to the Regulatory Review Process
- Office of Combination Products
Product Jurisdiction Program- Request for Designation - Memorandum of Understanding:
CBER/NINDS Interagency Working Group: 4th Year- PURPOSE: Provides an infrastructure to support information sharing between FDA/CBER and NIH/NINDS
- GOAL: To expedite translation of basic research involving promising biological therapies to well-designed clinical studies for the treatment of neurological disorders through enhanced information exchange.
- FORMAT: CBER and NINDS staff conduct monthly meetings to discuss regulations, policies, and statutory responsibilities, as well as address difficult questions and issues that confront development of new therapies.
- Laboratory-based, Research/Reviewer Model
Conduct research that supports FDA's Critical Path Initiative
Resources Important to the Regulatory Review Process
Cellular, Tissue and Gene Therpies Advisory Committee (CTGTAC)
http://www.fda.gov/oc/advisory/acbiologics.html
- The Committee reviews and evaluates available data relating to the safety, effectiveness, and appropriate use of biological response modifiers which are intended for use in the prevention and treatment of a broad spectrum of human diseases.
- Purpose: To provide the FDA with current, reliable scientific and medical guidance to facilitate regulatory decisions pertaining to cellular, tissues and gene transfer therapies regulated by CBER
Resources Important to the Regulatory Review Process
CBER-CDRH Cross Center Teams: Cardiovascular and Cartilage Repair Products
- Formed to address regulatory issues for products having both biological and device components.
- Identify emerging technologies (cell-scaffold constructs, cell delivery devices)
- Provide recommendations concerning develop-ment of regulatory policy and product-specific guidance/standards
- Facilitate coordinated / timely cross center review of submissions involving biological (cell, gene, tissue) and device components
Guidance Documents and Rule Making
- Provide enhanced flexibility, more readily updated.
- Are developed in consultation with all interested stakeholders- industry and the public.
- Enable the Agency to maintain currency with developing technological capabilities.
- Finalized rules are considered to carry the weight of regulation.
CRITICAL PATH INITIATIVE: The Goals
- Bring scientific advances to medical product development process (e.g. simulation models, validated biomarkers, new clinical trial designs).
- Stimulate development of robust applied research programs in critical path scientific areas, aim to develop techniques that remove specific obstacles encountered during product development.
- Modernize regulatory standards in order that they reflect the best available science, integrate FDA involvement.
Regulation of Cellular and Tissue-Based Products: Tissue Action Plan
- Provides a unified regulatory framework
- Provides greater flexibility intended to encourage innovation in the field of cellular therapies
- Provides a tiered regulatory approach with the level of regulation proportional to the degree of risk
- Risk determines level of regulation
- Lower Risk - Tissue Regulations Suffice: Section 361, PHS Act, 21 CFR Part 1271- Human Cells, Tissue and Cellular and Tissue-Based Products
- Higher Risk - Preapproval Required: Section 351, PHS Act (Biologic); Section 505 Food, Drug and Cosmetic Act (Drug), Investigational New Drug Requirements - 21 CFR Part 312.
Regulation of Cellular and Tissue-Based Products: Tissue Action Plan
- Novel biologic therapies comprised of, or derived from, stem cells will be regulated as human cells, tissues or cellular or tissue-based products: HCT/P's
21 CFR 1271.3(d)- (HCTP) means articles containing or consisting of human cells or tissues that are intended for implantation, transplantation, infusion, or transfer into a human recipient
Regulatory Framework Goals
- Prevent unwitting use of contaminated tissues with the potential for transmitting infectious disease
- Prevent improper handling or processing that might contaminate or damage tissues
- Ensure that clinical safety and effectiveness is demonstrated for cells and tissues that are highly processed, used for purposes other than re-placement, combined with non-tissue compon-ents, or that have systemic effects
Tissue Rules
- The Establishment Registration, Donor Eligibility and GTP final rules comprise 21 CFR Part 1271
- These three rules form the platform for regulation of all human cells, tissues, and cellular and tissue-based products (HCT/P's)
- For certain HCT/P's (361 HCT/P's), the new regulations will comprise the sole regulatory requirements
- For HCT/P's regulated as drugs, devices, and/or biological products, the new regulations will supplement GMP/QSR
Regulation of Stem Cell Therapies Under the Tissue Action Plan Framework
HCT/P's regulated solely under section 361 of the PHS Act (for pre-venting introduction, transmission and spread of communicable diseases) and 21 CFR Part 1271 ONLY if ALL FOUR of the following are met:
- Minimally manipulated: Relevant biologic characteristic(s) are not altered by processing.
- Homologous Use Only: The HCT/P performs the same basic function or functions in the recipient as in the donor
- Production of the HCT/P does not involve combination of cells with another article (with limited exceptions and on the condition that addition of the excepted article does not raise new clinical safety concerns)
- Does not have a systemic effect, is not dependent upon the metabolic activiity of living cells for primary function: exceptions for (a) autologous use, (b) first- or second- degree blood relatives, or (c) reproductive use.
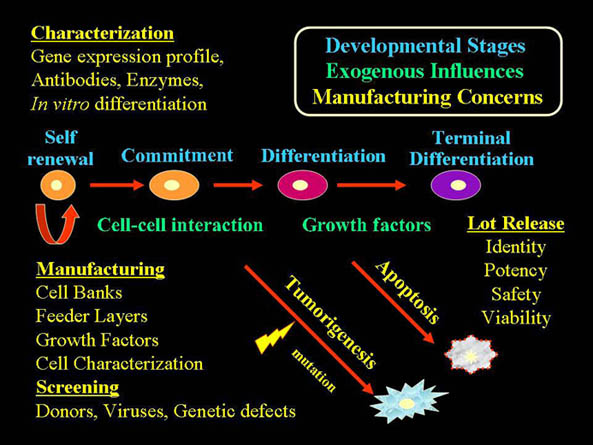
Stem Cells: Biological Characteristics Convey Both Therapeutic Promise and Regulatory Challenges
- Capacity for self-renewal, robust proliferative potential.
- Capable of differentiating into varied, disparate tissue phenotypes in response to appropriate biologic cues.
- Putative Plasticity / Transdifferentiation
ALL OF THE ABOVE!!!
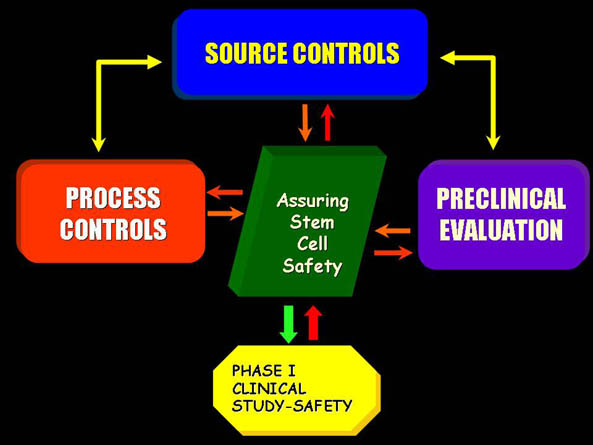
Developing a Stem Cell-Based Product: Source Controls
- Evaluating Human Stem Cell Sources
- Appropriate screening/testing of donor tissue for communicable disease is essential- 21 CFR 1271, Subpart C: Donor Eligibility Rule
- Consider implications of molecular genetic analysis
- Determine whether intrinsic safety concerns exist based on cell source (adult, fetal, embryonic)
- Develop and standardize criteria for accepting donor source materials to initiate production of a stem cell based investigational product
Developing a Stem Cell-Based Product: Process Controls
- Critical Manufacturing Process Controls
- Standardization and optimizatin of reagents and processing procedures
- Product characterization and development of acceptance criteria
- Controlling purity and impurities profiles of the final cellular product
- Establish specific characteristics to ensure product integrity
- Identify product parameters that anticipate adverse events
- Develop analytical approaches for evaluating proposed acceptance criteria for in process intermediates and final cellular product
Developing a Stem Cell-Based Product: Detailed Characterization
- Detailed characterization of stem cell-based products involves multi-parametric analytical testing:
- Morphologic evaluation
- Detection of phenotype-specific cell surface antigens
- Unique biochemical markers
- Gene and protein expression analysis(microarray and proteomics)
- cellular impurities profile assessment
- Biologic activity assay ≈ potency
- MHC/HLA expression predicting immunologic compatibility/anticipating immunogenicity
Developing a Stem Cell-Based Therapy: Assessing Potency
- Potency Assays: measure the relevant biological function / activity of the product. (Topic of Recent CTGT Advisory Committee Meeting: February 9, 2006)
- Development of stem cell potency bioassay
- Validate surrogate markers as predictive of intended biologic activity
- Specify parameters on which product dosing should be based (i.e., number of viable cells, activity, molecular marker)
Developing a Stem Cell-Based Product: Preclinical Evaluation
- Animal testing: Toxicological Assessment
- Comprehensive histological examination evidence for:
- Cell survival post transplantation
- Cell migration
- Cellular fate-plasticity: differentiation, transdifferentiation, fusion
- Tissue integration
- Tumorigenicity (hyperplastic or unregulated growth)
Human Embryonic Stem Cell Lines
- Issues Receiving Attention:
- Media used for culturing hES cells is routinely supplemented with bovine serum (concern over BSE/TSE, vCJD) as well as other animal-derived ancillary products.
- Characterization of therapies derived from hES cells as xenotransplantation products: use of irradiated murine embryonic fibroblast feeder layers.
- Published technical report in Nature Medicine: Human embryonic stem cells express a nonhuman immunogenic sialic acid (Neu5Gc).
- Karyotypic / genetic stability of long-term hES cell cultures
Developing a Stem Cell-Based Product: Additional Tasks to Facilitate Review
- Develop analytical tools and experimental techniques that enable assessments to be made concerning the comparability/degree of equivalence between stem cell-based products derived from different starting source materials
- For long term cultures of stem cell populations determine the impact of genetic instability observed on biologic properties pertinent to their utility as cellular products
- Develop new animal based or in vitro preclinical models to: (1) assess feasibility, (2) identify potential target tissues for toxicity/activity, (3) conduct dose exploration, (4) identify parameters to monitor during clinical investigation
Developing a Stem Cell-Based Product: Additional Tasks (cont)
- Develop non-invasive technologies with limits of detection that enable investigation of the fate of stem cell-based products following administration: (1) assess viability, (2) monitor additional differentiation (3) observe migration, (4) detect physiologic integration
- Evaulate approaches for the delivery of stem cell-based products:
- Catheter/needle injection systems-reliably and reproducibly deliver targeted number of viable cells
- Biocompatible cell scaffolds and matrices: tissue contstructs
- Encapsulation methodologies that prevent immunologic rejection while permitting release of bioactive materials from encapsulated cells (i.e. β-islets, insulin)
Regulatory Approach to Evaluating Human Stem Cell Therapies
- The review of Investigative New Drug Applications (IND's)that involve human stem cell products will be based on the best available science
- When appropriate, CBER will seek input from its relevant advisory committees
- To maximize flexibility, rules, and guidance documents will be integral components of the regulatory decision making process
- CBER encourages early interactions between itself and sponsors as necessary in order to facilitate an efficient and effective product review process
Helpful Hints
- When in doubt or unsure about an issue, seek Agency advice.
- For novel investigational products or the uninitiated sponsor, take advantage of the pre-IND meeting opportunity to seek Agency guidance and advice that reflects "current thinking".
- Don't delay addressing critical tasks until the 11th-hour.
- Consider your interaction with the Agency to be a partnership that will assist you in meeting regulatory requirements for demonstrating safety and efficacy.
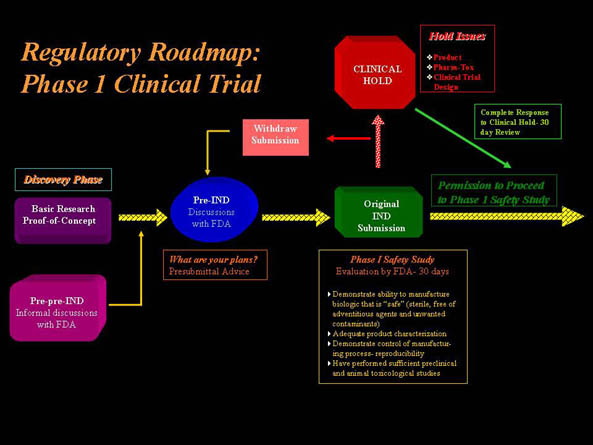
Selected Relevant Guidance Documents Supporting Regulatory Review of Stem Cell-Based Therapies
- TISSUE ACTION PLAN: FDA Approach to the Regulation of Cellular and Tissue-Based Products- http://www.fda.gov/cber/tissue
- Guidance for INdustry: IND;s - Approaches to Complying with cGMP During Phase 1- January 2006 http://www.fda.gov/cber/gdlns/indcgmp.pdf
- Draft Guidance for Reviewers: Intructions and Template for Chemistry, Manufacturing, and Control (CMC) Reviewers of Human Somatic Cell Therapy Investigational New Drug Applications (INDs)- 8/15/2003 http://www.fda.gov/cber/gdlns/cmcsomcell.pdf
- Final Rule: Eligibility Determination for Donors of Human Cells, Tissues, and Cellular and Tissue-Based Products (HCT/Ps)-5/20/2004 http://www.fda.gov/cber/gdlns/tissdonor.pdf
- Final Rule:Current Good Tissue Practice for Human Cell, Tissue, and Cellular and Tissue-Based Product Establishments; Inspection and Enforcement- 11/24/2004 http://www.fda.gov/cber/rules/gtp.pdf
- Guidance for Human Somatic Cell Therapy and Gene Therapy- 3/30/1988 http://www.fda.gov/cber/gdlns/somgene.pdf
Selected Relevant Guidance Documents Supporting Regulatory Review of Stem Cell-Based Therapies
- TISSUE ACTION PLAN: FDA Approach to the Regulation of Cellular and Tissue Based Products(cont) http://www.fda.gov/cber/tissue
- ICH Guidance on Viral Safety Evaluation of Biotechnology Products Derived From Cell Lines of Human or Animal Origin - 9/24/1998 http://www.fda.gov/cber/gdlns/virsafe.pdf
- Draft Points to Consider in the Characterization of Cell Lines Used to Produce Biologicals (1993)- 7/12/1993 http://www.fda.gov/cber/gdlns/ptccell.pdf
- Guidance for Industry: Guidance for Human Somatic Cell Therapy and Gene Therapy - 3/30/1998 http://www.fda.gov/cber/gdlns/somgene.pdf
- Guidance For the Submission of Chemistry, Manufacturing and Controls Information and Establishment Description for Autologous Somatic Cell Therapy Products - 1/10/1997 http://www.fda.gov/cber/gdlns/xvcmc.txt
Contacting the Center for Biologics
CBER CONTACT INFORMATION
- Phone: 1-800-835-4709(Within U.S.); 301-827-1800 (Local or Outside U.S.)
- INTERNET: http://www.fda.gov/cber
- Send email to:
- Consumers- Health Care Professionals: OCTMA@CBER.FDA.GOV
- Manufacturers- Regulated Industry:MATT@CBER.FDA.GOV
- CBER Regulated and Guidance Documents on the internet at:http://www.fda.gov/cber/guidelines.htm