


Guidance for Industry
Gene Therapy Clinical Trials – Observing Subjects for Delayed Adverse Events
Additional copies of this guidance are available from the Office of Communication, Training and Manufacturers Assistance (HFM-40), 1401 Rockville Pike, Suite 200N, Rockville, MD 20852-1448, or by calling 1-800-835-4709 or 301-827-1800, or from the Internet at http://www.fda.gov/cber/guidelines.htm.
For questions on the content of this guidance, contact the Office of Cellular, Tissues, and Gene Therapies at 301-827-5102.
U.S. Department of Health and Human Services
Food and Drug Administration
Center for Biologics Evaluation and Research
November 2006
- INTRODUCTION
- BACKGROUND
- Potential Risks of Delayed Adverse Events Following Exposure to Gene Transfer Technology
- Previous FDA Recommendations
- Concerns Raised by the Gene Therapy Community
- DEFINITIONS AND ABBREVIATIONS
- PRECLINICAL DATA USED FOR ASSESSMENT OF DELAYED RISKS IN GENE THERAPY CLINICAL TRIALS
- Criteria to Assess Potential Delayed Risks of Gene Therapy
- Considerations for Preclinical Study Design to Assess Vector Biodistribution and Persistence
- Animal Study Design
- Tissue Collection and Analysis
- Other Considerations
- Vector Integration Potential and Reactivation as Risks for Delayed Adverse Events
- RECOMMENDATIONS FOR PROTOCOLS FOR LONG-TERM FOLLOW-UP OBSERVATIONS: CLINICAL CONSIDERATIONS
- Decision to Conduct Long-term Follow-up Observations
- Suitability of Clinical Trial Populations for Long-term Follow-up Observations
- Recommended Duration of Follow-up Observations
- Elements of Follow-up Observations
- Informed Consent in Trials Involving Long-term Follow-up Observations
- Special Considerations Regarding Integrating Vectors
- Data Collection
- Data Reporting
- Informed Consent in Trials Involving Retroviral Vectors
- REFERENCES
Contains Nonbinding Recommentions
Guidance for Industry
Gene Therapy Clinical Trials - Observing Subjects for Delayed Adverse Events
| This guidance represents the Food and Drug Administration’s (FDA’s) current thinking on this topic. It does not create or confer any rights for or on any person and does not operate to bind FDA or the public. You can use an alternative approach if the approach satisfies the requirements of the applicable statutes and regulations. If you want to discuss an alternative approach, contact the appropriate FDA staff. If you cannot identify the appropriate FDA staff, call the appropriate number listed on the title page of this guidance. |
- INTRODUCTION
- BACKGROUND
- Potential Risks of Delayed Adverse Events Following Exposure to Gene Transfer Technology
- Previous FDA Recommendations
- Concerns Raised by the Gene Therapy Community
- Not all gene therapy products present the same risks of delayed adverse events. Uniform recommendations for long-term follow-up for all gene therapy products did not take product characteristics into account.
- Some study subjects appear unsuitable for meaningful long-term follow-up observations because of high short-term mortality, poor general health, or exposure to mutagenic agents.
- Our recommendations regarding the duration and design of long-term follow-up have not been sufficiently specific.
- DEFINITIONS AND ABBREVIATIONS
- PRECLINICAL DATA USED FOR ASSESSMENT OF DELAYED RISKS IN GENE THERAPY CLINICAL TRIALS
- Criteria to Assess Potential Delayed Risks of Gene Therapy
- Question 1: “Is your gene therapy product used only for ex vivo modification of cells?”
- Question 2: “Do preclinical study results show persistence of vector sequences?”
- Question 3: “Are vector sequences integrated?”
- Question 4: “Does the vector have potential for latency and reactivation?”
- Your product is a plasmid and the similar product is also a plasmid, but has different coding sequences for the proposed therapeutic gene product. The similar product has been used in preclinical and clinical studies, administered by an identical route and in an identical final formulation to that proposed in the prospective studies. Reference to a published study demonstrating lack of persistence of the vector for the similar product may adequately address concerns regarding the persistence of the proposed vector.
- Your proposed product and the similar product differ only with respect to route of administration. The similar product was administered into tumors (intratumorally). The proposed product is to be given intravenously. There is a published study demonstrating the lack of persistence of the vector when administered intratumorally. The data from the studies with the similar product are not sufficiently relevant, since there was no intended systemic exposure to the product. Thus, there is insufficient similarity to conclude that long-term follow-up observations are not necessary to mitigate long-term risks to subjects. In the absence of relevant data from a study involving a similar product, we recommend that you assess the risk of vector persistence in a preclinical study with the proposed product administered by the intravenous route.
- A preclinical toxicology study indicates that expression of the transgene is associated with delayed toxicity.
- The transgene provides functional replacement of a host gene; the transgene product is potentially immunogenic.
- Data collected in your short-term clinical study indicate vector persistence, even though data from your preclinical studies suggested that the vector did not persist.
- Considerations for Preclinical Study Design to Assess Vector Biodistribution and Persistence
- Animal Study Design
- Use the product in the final formulation proposed for the clinical study because changes in the final formulation may alter biodistribution patterns.
- Use both genders or justify the use of a single gender.
- Use at least 5 animals per gender per group per sacrifice time point for rodents, and between 3-5 animals per gender per group per sacrifice time point for nonrodents.
- Consider factors in the study design that might influence or compromise the vector distribution and/or persistence such as the animal’s age and physiologic condition.
- Use the intended clinical route of vector administration if possible.
- Assess vector biodistribution in a vehicle control group and a group of animals that receives the MFD or clinically relevant dose (defined in Section III). Studies at additional dose levels might provide dose-dependent information.
- Include appropriate safety endpoints in your biodistribution study in order to assess any potential correlation between vector presence/persistence and adverse findings if safety endpoints have not been evaluated already in a separate toxicity study using the same animal model. These safety endpoints should include clinical observations, body weights, clinical pathology, gross organ pathology, and histopathology.
- Include several sacrifice intervals to characterize the kinetics of vector distribution and persistence. We recommend
sacrifice at the expected time of peak vector detection and at several later time points to evaluate clearance of vector sequences from tissues.
- Tissue Collection and Analysis
- Sample and analyze the following panel of tissues, at a minimum: blood, injection site(s), gonads, brain, liver, kidneys, lung, heart, and spleen. Consider other tissues for evaluation, depending on the vector type and the transgene, as well as the route of administration (e.g., draining lymph nodes and contralateral sites for subcutaneous/intramuscular injection, bone marrow, eyes, etc.).
- Choose a method for tissue collection that avoids the potential for contamination among different tissue samples.
- Use a quantitative, sensitive PCR assay to analyze the samples for vector sequences. You should submit data to your IND to demonstrate that your assay methodology is capable of specifically detecting vector sequence in both animal and human tissues. We recognize that PCR technology is constantly changing, and encourage you to discuss the assay methology with us before initiating sample analysis. Current recommendations include the following:
- The assay should have a demonstrated limit of quantitation of ≤50 copies of vector/1 µg genomic DNA, so that your assay can detect this limit with 95% confidence.
- Use a minimum of three samples per tissue. One sample of each tissue should include a spike of control DNA, including a known amount of the vector sequences, in order to assess the adequacy of the PCR assay reaction. The spike control will determine the specified PCR assay sensitivity.
- Provide a rationale for the number of replicates for testing per tissue, taking into account the size of the sample relative to the tissue you are testing.
- Other Considerations
- Vector Integration Potential and Reactivation as Risks for Delayed Adverse Events
- Submit data to your IND from preclinical studies to assess vector persistence in an appropriate model. As stated in Section IV.B.3, we encourage you to discuss with FDA your study design before starting the trial.
- If the vector is not persistent, the predicted risk of delayed adverse events would be low. Long-term follow-up observations would be at your discretion.
- If the vector is persistent, we recommend that you perform preclinical studies to assess vector integration, as well as the potential for vector latency and reactivation.
- If the studies show no evidence for persistence due to integration of the genetic material or development of latency, the predicted risk of delayed adverse events would be low. Long-term follow-up observations would be at your discretion.
- If the studies show no evidence for integration of the genetic material but studies for latency and reactivation are inconclusive, cannot be performed, or show evidence of latency and/or reactivation, the predicted risk of delayed adverse events is indeterminate. We would require long-term follow-up observations.
- If preclinical studies of vector integration are not feasible, if the genetic material integrates, or if the vector is shown to persist in a latent state that may be reactivated, the risk of delayed adverse events is high or unknown, and long-term follow-up observations in study subjects are warranted.
- If vector integration studies are not performed, we recommend that you provide other evidence to support an assessment that your vector does not pose high risks of delayed adverse events, including the following:
- A discussion of why vector integration studies were not performed.
- The evidence supporting your assessment of the risk of delayed adverse events posed by your product.
- RECOMMENDATIONS FOR PROTOCOLS FOR LONG-TERM FOLLOW-UP OBSERVATIONS: CLINICAL CONSIDERATIONS
- Decision to Conduct Long-term Follow-up Observations
- The answers to the questions posed in Section IV, Figure 1. “Framework to Assess the Risk of Gene Therapy-Related Delayed Adverse Events” lead you to decide that the risks associated with your product are high or uncertain.
- The information about your product, taken as a whole, shows that long-term follow-up observations would mitigate the risks to human subjects. For examples of such circumstances please refer to the final paragraphs in Section IV.A.
- Suitability of Clinical Trial Populations for Long-term Follow-up Observations
- Recommended Duration of Follow-up Observations
- The observed duration of in vivo vector persistence;
- The observed duration of in vivo transgene expression;
- The prior, concomitant, and post gene therapy exposures of the study population;
- The expected survival rates in the study population; and
- Other factors that may be relevant to the feasibility and scientific value of conducting long-term follow-up observations.
- Elements of Follow-up Observations
- Implement methods for detection of gene therapy-related delayed adverse events;
- Assure that investigators maintain in the case history a detailed record of all exposures to mutagenic agents and other medicinal products and have ready access to information about their adverse event profiles;
- Design a plan for scheduled visits with a health care provider to elicit and record new findings for each study subject, including history, physical examination, or laboratory testing at minimum intervals of one year;
- Establish a method for investigators to record the emergence of new clinical conditions, including:
- New malignancy(ies)
- New incidence or exacerbation of a pre-existing neurologic disorder
- New incidence or exacerbation of a prior rheumatologic or other autoimmune disorder
- New incidence of a hematologic disorder; and
- Design a plan to elicit the cooperation of study subjects and their health care providers in reporting delayed adverse events, including unexpected illness and hospitalization.
- Contact subjects at a minimum of once a year. At your discretion, unless the long-term follow-up observation plan provides for additional, specific screening, you may arrange to contact subjects by telephone or written questionnaire rather than by office visits with a health care provider.
- Continue appropriate follow-up methods as indicated by previous test results. For example, it would be appropriate to monitor for vector sequences in subjects who had previous test results demonstrating vector persistence.
- Detection of Adverse Events: To facilitate detection of delayed adverse events, we recommend that the protocol identify suitable health care professionals whose observations would be used in the assessment of the occurrence of adverse events in the study population. Suitable health care professionals might include physicians, physician’s assistants, and nurse practitioners who were not otherwise associated with the clinical trial. You may arrange to have such individuals notified to provide prompt reports of adverse events to the investigators.
- IND Safety Reports: You must follow applicable reporting requirements outlined in 21 CFR 312.32 for adverse experiences associated with the use of the product. As the long-term follow-up observations proceed, you must also notify each participating investigator of any adverse experience associated with the use of the gene therapy product that is both serious and unexpected (21 CFR 312.32(c)(1)(i)(A)), as well as any new observations discovered by, or reported to, you (21 CFR 312.55(b)). In each IND Safety Report (required to be provided to nvestigators and FDA), you must identify all safety reports previously filed concerning a similar adverse experience, and analyze the significance of the adverse experience in light of the previous, similar reports (21 CFR 312.32(c)(1)(ii)). You must promptly investigate all safety information you receive (21 CFR 312.32(d)(1)). If the relationship of the adverse experience to the gene therapy product is uncertain, we may recommend that you perform additional investigations and revise your Informed Consent Document and Investigator Brochure to inform all study subjects of the risk of the adverse experiences. We may also request that investigators contact previously treated study subjects to inform them of the new risk.
- Annual Reports to the IND/Summary Information: While the IND is in effect and until long-term follow-up observations are concluded, you must file an annual report. In that report, submit information obtained during the previous year's clinical and nonclinical investigations, including, among other things, a summary of all IND safety reports submitted during the past year, and a narrative or tabular summary showing the most frequent and most serious adverse experiences by body system (21 CFR 312.33(b)(1) and (2)).
- Amendments to Your Clinical Protocol: If clinical data suggest that your product is not associated with delayed risks, you may want to consider changing the clinical protocol regarding long-term follow-up of study subjects. However, before implementation of this change, you must submit to FDA a protocol amendment to your IND indicating the relevant changes (21 CFR 312.30(b)(1), (d), and (e)).
- Scheduled Physical Examinations: We recommend that long-term follow-up observations include scheduled physical examinations performed by a health care professional at least once a year during the first five years, unless the assessed risks associated with your protocol indicate that they should be done more frequently. For example, if a subject exposed to your product or an analogous product develops a rapidly progressive, potentially reversible delayed adverse event, and there is a reasonable possibility that the event may have been caused by the product, it may then become advisable to perform observations on a semi-annual or quarterly basis. Such periodic evaluation should include a brief history and focused examination designed to determine whether there is any evidence of emergence of clinically important adverse events. Appropriate laboratory evaluations, such as a hematology profile, should be included with the periodic physical examination. Long-term follow-up observations are intended for study purposes only, not to provide evaluation and treatment of health care problems that are not associated with the use of the product.
- Vector Sequences: During long-term follow-up, we recommend that you test study subjects at least annually for persistent vector sequences until they become undetectable. The assay should be sufficiently sensitive to detect vector sequences. We recommend that you sample the likely population of transduced cells without being overly invasive (e.g., peripheral blood is a suitable sample to test for presence of hematopoietic stem cells, rather than bone marrow biopsy). In those cases where the transduced cell population may require an invasive procedure, we recommend that you consider, instead, measuring a surrogate that may indicate vector persistence (e.g., the level of transgene product or some clinical effect). Data demonstrating the lack of detectable vector may provide a rationale to revise the long-term follow-up elements of your study as an amendment to your IND. In any such protocol amendment, include an assessment of risks associated with your product and an evaluation of the impact of the waning persistence of the vector on those risks (21 CFR 312.30(b), (d)(2)).
- Informed Consent in Trials Involving Long-term Follow-up Observations
- Special Considerations Regarding Integrating Vectors
- Data Collection
- The choice of method to assess the pattern of vector integration sites should be based upon data with appropriate positive and negative controls (i.e., target cells with a known number and sites of vector copies integrated vs. target cells with no vector integrants). Studies should be performed to provide information about the assay sensitivity, specificity, and reproducibility.
- We recommend that you perform an analysis to assess the pattern of vector integration sites if at least 1% cells in the surrogate sample are positive for vector sequences by PCR. As an alternative, you may base the decision to analyze for clonality of vector integration sites on an evaluation of the sensitivity of the assay system used to detect clonality.
- We recommend that you test for vector sequences by PCR in subject surrogate samples obtained at intervals of no greater than six months for the first five years and then no greater than yearly for the next ten years, or until such time that no vector sequences are detectable in the surrogate sample.
- We recommend that you perform an analysis to determine the site of vector integration if the analysis of a subject’s surrogate cells suggests a predominant clone (e.g., oligoclonal pattern of vector insertions) or monoclonality. In addition, if you detect a predominant integration site, test for persistence by performing another analysis for clonality no more than three months later.
- When the nucleotide sequence adjacent to the site of the vector integration has been determined, we recommend that you compare the identified integration site sequence with known human sequences in the human genome database and other databases that document oncogenes to determine whether the identified sequences are known to be associated with any human cancers.
- While we recognize that oligoclonality or even monoclonality itself will not a priori result in a malignancy (Refs. 15 and 16), we also recognize that these changes increase the risk of a malignancy, and therefore, we recommend that you institute a plan to monitor the subject closely for signs of malignancy if any of the following conditions pertain:
- Persistent monoclonality;
- Clonal expansion (e.g., the per cent cells positive for a particular vector integration site is shown to increase over multiple timepoints); or
- Evidence of vector integration near or within a locus known to have oncogenic activity.
- To screen for specific disease entities, we recommend that you use established methods and/or seek advice from clinicians with expertise in screening for the health care risks to which, according to your evidence, your subjects may be exposed.
- Data Reporting
- Informed Consent in Trials Involving Retroviral Vectors
- Description of study agent - The study involves giving a person some cells that have been changed by a retroviral vector. A retroviral vector is a virus that can insert genetic material into cells.
- Mechanism of action for retroviral vectors - When retroviral vectors enter a normal cell in the body, the deoxyribonucleic acid (DNA) of the vector inserts itself into the normal DNA in that cell. This process is called DNA integration.
- Effect of DNA integration - Most DNA integration is expected to cause no harm to the cell or to the patient. However, there is a chance that DNA integration might result in abnormal activity of other genes. In most cases, this effect will have no health consequences.
- Discussion of cancer occurring in animal studies - In some cases, abnormal activity of a normal gene may cause an uncontrolled growth of the cell that sometimes results in a cancer. This type of event has occurred in animal studies in which retroviral vector DNA integration appeared to cause cancers in mice and monkeys.
- Discussion of delayed adverse event, leukemia-like malignancy, occurring in human studies - It is important that you know about some cancers that occurred in another gene therapy research study. The study, conducted in France, involved a disease called X-linked Severe Combined Immunodeficiency (SCID). Years after receiving cells that were modified by a retroviral vector, a significant number of the children in this small study developed a leukemia-like malignant disease (cancer). At least one child died from the cancer. A group of experts in this field studied the results from tests performed on these children’s blood cells. They concluded that the leukemia-like malignancy was caused by the retroviral vector DNA. However, most of the children with X-linked SCID who have received experimental gene therapy have not been found to have a leukemia-like disease at this time. Although they appear healthy, we still do not know whether they, too, will develop a malignant growth.
- Risk of malignancy for this study - We do not know if the retroviral vector used in this protocol might cause a new malignancy. However, you should be aware that the DNA contained in retroviral vectors will integrate into your DNA and that under some circumstances, this has been known to cause malignant (cancerous) growth months to years later.
- REFERENCES
- FDA Guidance for Industry: Supplemental Guidance on Testing for Replication Competent Retrovirus in Retroviral Vector Based Gene Therapy Products and During Follow-up of Patients in Clinical Trials Using Retroviral Vectors, November 2006 (http://www.fda.gov/cber/gdlns/retrogt1000.pdf).
- Donahue, R.E., et al., Helper virus induced T cell lymphoma in nonhuman primates after retroviral mediated gene transfer. Journal of Experimental Medicine 1992; 176:1125-1135.
- Nyberg, K., et al., Workshop on long-term follow-up of participants in human gene transfer research. Molecular Therapy 2004; 10(6):976-980.
- FDA Guidance for Industry: Guidance for Human Somatic Cell Therapy and Gene Therapy, March 1998 (http://www.fda.gov/cber/gdlns/somgene.pdf).
- Bauer, S., Current FDA approach for preclinical vector biodistribution studies, March 12, 1999: Recombinant DNA Advisory Committee Meeting (http://www4.od.nih.gov/oba/rac/minutes/3-99RAC.htm#IX).
- Shayakhmetov, D.M., et al., A high-capacity, capsid-modified hybrid adenovirus/adeno-associated virus vector for stable transduction of human hematopoietic cells. Journal of Virology 2002; 76(3):1135-1143.
- Goncalves, M.A., et al., Stable transduction of large DNA by high-capacity adeno-associated virus/adenovirus hybrid vectors. Virology 2004; 321(2):287-96.
- Picard-Maureau, M., et al., Foamy virus—adenovirus hybrid vectors. Gene Therapy 2004; 11(8):722-28.
- Yant, S.R., et al., Transposition from a gutless adeno-transposon vector stabilizes transgene expression in vivo. Nature Biotechnology 2002; 20(10):999-1005.
- Wang, Z., et al., Detection of integretation of plasmid DNA into host genomic DNA following intramuscular injection and electroporation. Gene Therapy 2004; 11(8):711-21.
- Li, Z., et al., Murine leukemia induced by retroviral gene marking. Science 2002; 296:497.
- Modlich, U., et al., Leukemias following retroviral transfer of multidrug resistance 1 (MDR1) are driven by combinatorial insertional mutagenesis. Blood 2005; 105(11):4235.
- Hacein-Bey-Abina, S., et al., LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003; 302(5644): 415-9.
- Couzin, J., Kaiser, J., As Gelsinger Case Ends, Gene Therapy Suffers Another Blow. Science 2005; 307:1028.
- Ott M.G., et al., Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nature Medicine 2006; 12(4):401-9.
- Schmidt M., et al., Clonality analysis after retroviral-mediated gene transfer to CD34+ cells from the cord blood of ADA-deficient SCID neonates. Nature Medicine 2003; 9(4):463-68.
This guidance provides to you, sponsors of gene therapy studies, recommendations regarding the design of studies to include the collection of data on delayed adverse events in subjects who have been exposed to investigational gene therapy products. We, FDA, are providing: (1) recommended methods to assess the risk of gene therapy-related delayed adverse events following exposure to investigational gene therapy products, (2) recommended methods to determine the likelihood that long-term follow-up observations on study subjects will provide scientifically meaningful information, and (3) specific advice regarding the duration and design of long-term follow-up observations1. When a gene therapy clinical trial presents long-term risks to human subjects, a gene therapy clinical trial must provide for long-term follow-up observations in order to mitigate those risks. Without such long-term follow-up observations, the study would expose the subjects to an unreasonable and significant risk of illness or injury (21 Code of Federal Regulations (CFR) 312.42(b)(1)(i) and (b)(2)(i)).
Exposure to gene transfer technology means any exposure to gene therapy products or to cells or tissue that has been transduced with gene therapy products ex vivo by any route of administration. Except as noted below, this guidance applies to all subjects in clinical studies using gene transfer technology. The recommendations in this guidance are limited to the performance of long-term observations for evidence of delayed adverse events, i.e., adverse events that occur more than one year after exposure to the investigational gene therapy product.
This guidance finalizes the draft guidance entitled “Guidance for Industry: Gene Therapy Clinical Trials – Observing Participants for Delayed Adverse Events” dated August 2005. This guidance also supplements the recommendations for study subject long-term follow-up in the “Guidance for Industry: Supplemental Guidance on Testing for Replication Competent Retrovirus in Retroviral Vector Based Gene Therapy Products and During Follow-up of Patients in Clinical Trials Using Retroviral Vectors” (Retroviral Vector guidance), dated November 2006 (Ref. 1).
FDA’s guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidances describe the FDA’s current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in FDA’s guidances means that something is suggested or recommended, but not required.
Study subjects exposed to gene transfer technology may be at risk of delayed adverse events as a consequence of persistent biological activity of the genetic material or other components of the products used to carry the genetic material. The persistent biological activity may be necessary for the product to provide a continuing clinical benefit. However, persistent biological activity could have adverse effects upon normal cell function, placing subjects at risk for development of adverse events, some of which may be delayed by months or years.
Factors likely to increase the risk of delayed adverse events following exposure to gene transfer technology include persistence of the viral vector, integration of genetic material into the host genome, prolonged expression of the transgene, and altered expression of the host’s genes. Persistence of the viral vector, sometimes associated with latency, could permit continued expression of the gene or delayed effects of viral infection. Integration of genetic material from a viral vector into the host cell genomic DNA raises the risk of malignant transformation (see Section V.F for a discussion of risks of malignancy associated with retroviral vectors). Prolonged expression of the transgene may also be associated with long-term risks resulting from unregulated cell growth and malignant transformation, autoimmune-like reaction to self antigens, and unpredictable adverse events. Altered expression of the host genes could also result in unpredictable and undesirable biologic events.
We previously issued a guidance related to retroviral vector-mediated gene therapy (Ref. 1). We considered retroviruses to carry the highest known risk because of a reported case of new malignancy associated with a preclinical gene therapy study following exposure to cells transduced by a retroviral vector (Ref. 2), and therefore included in that guidance specific recommendations on performing long-term observations of subjects in trials of retroviral-mediated gene therapies.
We then sought additional information regarding gene-therapy related delayed adverse events following exposure to other gene-therapy products. We convened three separate meetings of our Biological Response Modifiers Advisory Committee (BRMAC) to solicit advice about long-term risks to subjects in gene therapy clinical trials exposed to other gene therapy products. The BRMAC meetings were held on November 17, 2000; April 6, 2001; and October 24, 20012. Since 2001, and after reviewing BRMAC’s recommendations, we have advised sponsors of studies involving gene transfer technology to submit to us their plans for long-term follow-up observations. We typically advised sponsors to observe subjects for potential gene therapy-related delayed adverse events for a 15 year period, and to include a minimum of five years of annual examinations, followed by ten years of annual queries, either in person or by questionnaire, of study subjects.
Members of the gene therapy community asked that the issue of long-term follow-up following exposure to gene transfer technology be discussed in a public forum. Accordingly, in June 2004 a public workshop was held in association with the annual meeting of the American Society of Gene Therapy (ASGT). The workshop was entitled “Long-Term Follow-Up of Participants in Human Gene Transfer Research” and was co-sponsored by the ASGT, Biotechnology Industry Organization (BIO), CBER, the NIH Office of Biotechnology Activities (OBA), and Pharmaceutical Research and Manufacturers of America (PhRMA). The workshop included a forum in which invited speakers discussed the challenges associated with long-term follow-up of subjects in gene therapy clinical studies. The workshop organizers published a summary of the discussion (Ref. 3).
Key issues identified by workshop participants include the following:
These issues are addressed in Sections IV and V of this guidance.
The following definitions apply to this guidance:
Gene therapy products:
All products that mediate their effects by transcription and/or translation of transferred genetic material and/or by integrating into the host
genome and that are administered as nucleic acids, viruses, or genetically engineered microorganisms. The products may be used to modify cells
in vivo or transferred to cells ex vivo prior to administration to the recipient.
Gene transfer:
The transfer of genetic material into a cell.
Gene transfer system:
The combination of the vector, regulating elements, vector formulation, and the route and method of vector delivery.
Gene transfer technology:
The use of genetic material either alone or in a suitable transfer medium, such as lipids, viruses, or other microorganisms, to mediate an effect
by transcription, translation, or integration into the host genome or any combination of these processes. Exposure to gene transfer technology may
result from direct administration of the product to a study subject or through use of cells or tissues exposed to such products ex vivo prior to
administration to a study subject.
IND:
Investigational New Drug Application, as described in 21 CFR Part 312.
Integration (of DNA):
The process whereby exogenous DNA sequences become incorporated into a genome.
Latency (of a viral infection):
A period of time during which a virus is present in the host without producing overt clinical symptoms.
Long-term follow-up observations:
Long-term follow-up observations are extended assessments that continue some of the scheduled observations of a customary clinical trial.
Long-term follow-up observations are an integral portion of the study of investigational products, such as gene therapy, that are considered
to present a high risk of producing delayed adverse events.
Maximum feasible dose (MFD) (in preclinical studies):
The highest dose that can be administered to a non-human animal. Limitations may be due to animal size, administration site, or product
characteristics. The MFD may not be equivalent to the clinically relevant dose.
Persistence:
With respect to transferred genetic material, the continued presence of genetic sequences in the host after acute exposure to a transfecting agent,
whether due to integration of the genetic sequence into the host genome or to latent infection with the viral vector bearing the genetic sequence.
Preclinical Study:
An investigational study performed in non-human animals or in isolated cells or tissue from humans or other animals. Preclinical studies may be
performed prior to or during clinical studies.
Reactivation (of a viral infection):
The re-emergence of a symptomatic or asymptomatic viral infection following a period of latency.
Transgene:
An exogenous gene that is introduced into a host cell.
Vector Sequences:
Refers to specific sequences of nucleotides, either DNA or RNA, that have been introduced into a gene therapy vector. The sequence includes all
components of the gene therapy vector, the vector backbone, transgene(s), and regulatory elements.
Viral Vector:
A virus that has been modified to transfer genetic material.
We generally will not require long-term follow-up observations following exposure to gene transfer technology when the risk of delayed adverse events is low. To assess the risk related to your product, we recommend that you use available preclinical and clinical evidence. To assess the risks of delayed adverse events, you may use current information about your product and similar products based on studies that you and others have performed. As more data accumulates, it is important to reassess the risk to your subjects and, if appropriate, revise your protocol as it relates to long-term follow-up observations.
We consider the assessment of risks to be a continuous process. New information may support the need for long-term follow-up observations or the revision of an existing study. For example, if recently reported evidence suggests a newly identified risk associated with your product or similar products, long-term follow-up observations may be necessary to mitigate long-term risks to subjects receiving these vectors. Similarly, if sufficient data accumulate to suggest that your product is not associated with delayed risks, it may be appropriate to reduce or eliminate provisions for long-term follow-up observations.
Pertinent previous preclinical and clinical experience with your product or similar products is highly relevant in the assessment of delayed adverse events. Experience with products in the same vector class, administered by a similar route, and given for the same clinical indication may contribute helpful information.
We recommend you refer to the series of questions in Figure 1, “Framework to Assess the Risk of Gene Therapy-Related Delayed Adverse Events” to help you assess the level of risk. When the risk of delayed adverse events is low based on your answers to these questions, a plan for long-term follow-up observations may not be necessary to mitigate risks to subjects. Evidence from preclinical studies will help you answer questions 1 – 3. Include all of the primary data relevant to the assessment of the risk of delayed events when you submit your IND to FDA (see 21 CFR 312.23(a)(8), (10)(iv), (11)).
We suggest you use the framework in Figure 1 by answering the questions in sequence as follows:
If the answer is “no,” go to Question 2. If the answer is “yes,” go to Questions 3 and 4.
If the answer is “no,” the risk of gene therapy-related delayed adverse events is low, and long-term follow-up observations may not be needed. If the answer is “yes,” go to questions 3 and 4.
If it is unknown whether your vector persists, for the purpose of assessing risk, we recommend that you either assume that it does persist, or perform a preclinical study to assay for vector persistence in a relevant animal species. Please refer to Section IV.B, “Considerations for Preclinical Study Design to Assess Vector Biodistribution and Persistence,” for help with preclinical trial design and details on the use and expected sensitivity of polymerase chain reaction (PCR) assay for biodistribution studies. In assays performed after the final administration of vector, persistence is indicated by detectable levels of vector sequences above the threshold level in the PCR assay and absence of an apparent downward trend over several time points. In contrast, persistence is unlikely if you cannot detect vector sequences with a sensitive PCR assay or if the assay for vector sequences demonstrates a downward trend over time. We encourage you to consult with OCTGT, CBER for specific advice about determination of persistence and biodistribution in your test system.
If the answer is “no,” go to question 4. If the answer is “yes,” we would require that clinical protocols with the product include clinical long-term follow-up observations.
If the answer is “no,” the risk is low that exposure to your gene transfer technology will be followed by gene therapy-related delayed adverse events. Long-term follow-up observations may not be needed. If the answer is “yes,” we would require that all your clinical protocols with the product include clinical long-term follow-up observations.
Laboratory and preclinical evidence of the low risk of delayed adverse events following exposure to a similar product may show that long-term follow-up observations are not needed. If you provide data from a similar product, we can assess the relevance to your product if you provide a clear explanation.
We provide the following two examples:
If you believe you have evidence from studies on a similar product that is adequate to support conclusions that the vector is unlikely to persist in human hosts and that the vector’s DNA does not integrate into the human genome, you may decide to submit a clinical protocol that does not provide for long-term follow-up observations. We will review such submissions and, if we disagree based upon our review of your submission or other additional information, we may conclude that long-term follow-up observations for delayed adverse events are necessary to mitigate long-term risks, and that without long-term follow-up observations, the study presents an unreasonable and significant risk to study subjects (21 CFR 312.42(b)(1)(i) and (b)(2)(i)).
We provide the following examples of evidence that might cause us to require you to perform long-term follow-up observations for delayed adverse events:
Figure 1. Framework to Assess the Risk of Gene Therapy-Related Delayed Adverse Events.
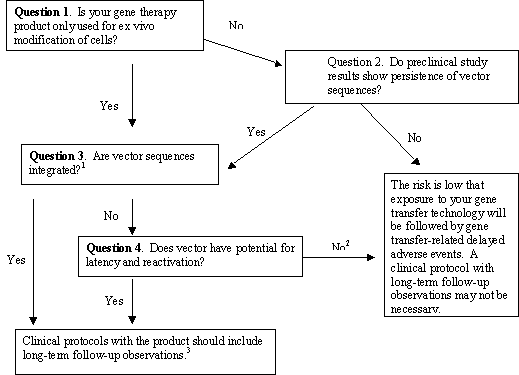
1If you have evidence that suggests that the vector may integrate or if the vector was intentionally designed to facilitate integration (please refer to Table 1, Section IV.C), the answer is “yes.” If you have no evidence regarding integration, we recommend that you include preclinical study in your development plan to address this question.
2If you or others identify an increased risk of delayed adverse events from persistent gene expression or from exposure to your product based on additional information reported after your protocol is accepted, you should plan to perform long-term follow-up observations even if the answer to these questions is “No”. See Section IV.A of the text for examples.
3See Section V of the text for recommendations on how to perform clinical long-term follow-up observations.
As discussed in Section IV.A, vector persistence heightens the risk of delayed adverse events following exposure to gene transfer technology. Indeed, the longer the vector persists, the greater the duration and degree of risk of delayed adverse events. We recommend that you perform preclinical biodistribution studies using methods that are shown to be sensitive and quantitative to detect vector sequences. Such studies would be designed to determine the distribution of your vector in nontarget tissues and the persistence of the vector in both nontarget and target tissues following direct in vivo administration of the vector product. If possible and applicable, we recommend that the studies employ an animal species that permits vector transduction and/or vector replication and that the animal species be biologically responsive to the specific transgene of interest (Ref. 4). The duration of the preclinical studies will vary, depending on the animal model employed. Projections of delayed adverse reactions in human subjects may be derived from assessment of data from appropriate long-term observational studies in animals, when possible.
A biodistribution study in animals can be performed either as a separate study or as a component of a toxicology study. Consider the following points in your animal study design to permit evaluation of vector localization and persistence (Ref. 5).
We encourage you to discuss with FDA your study design before starting the trial to ensure that the trial will adequately assess both biodistribution and vector persistence. There are many variables that will affect the outcome and interpretation of the in vivo assessment of each vector type.
Three gene therapy vectors currently under study (i.e., Gammaretrovirus, Lentivirus, and Herpesvirus) possess characteristics that we consider to pose high risks of delayed adverse events. Accordingly, we believe that clinical long-term follow-up observational studies would be necessary to mitigate long-term risks to subjects receiving these vectors. Gammaretrovirus and Lentivirus have a documented ability to integrate and Herpesvirus has a documented potential for latency and reactivation. In this section, we discuss those risks and the relatively low risks associated with gene transfer technology with vectors that lack those properties.
Most vectors used in gene therapy clinical trials can be categorized according to their propensity to integrate into host cell DNA. Please refer to Table 1, “Integration Properties of Current Commonly Used Gene Therapy Vectors in Clinical Trials.” As shown in Table 1 and reflected in the answer to question 3 in Figure 1, “Framework to Assess the Risk of Gene Therapy-Related Delayed Adverse Events,” vectors that have a potential to integrate present sufficient risk that long-term follow-up observations are necessary to mitigate long-term risks to subjects receiving these vectors.
Because of its potential for latency and reactivation, a Herpes virus-based gene transfer vector also presents a risk of delayed adverse events related to its use as a vector in gene therapy products. During latency, the virus and its gene products remain inactive. Reactivation may be delayed for months or years following initial exposure.
We are aware that the potential of vectors to integrate may be modified to increase their utility as gene therapy agents. For example, an adenovirus vector can be modified to induce integration of its DNA (Refs. 5-9). Another example would be changes in the methods used to introduce plasmid DNA vectors into cells that result in higher integration frequencies (Ref. 10). In those cases where a modification of the gene therapy system may have altered the persistence or integration properties, we recommend that you take one of the following actions:
Plasmids, poxvirus, adenovirus, and adeno-associated virus-based vectors (AAV) are vectors that do not have a propensity to integrate or reactivate following latency and, in the absence of evidence to the contrary, present a low risk of gene therapy-related delayed adverse events. However, even if your vector has a low propensity to integrate or reactivate, preclinical or clinical data showing persistence of the vector raise concerns about a risk of delayed adverse events, and follow-up observations would be necessary to mitigate long-term risks to subjects receiving these vectors. For example, if an AAV vector is shown to have persistent transgene expression, the risk of a delayed aberrant immune response should be considered because of the potential for autoimmune phenomena.
We also note that some vectors currently considered to pose delayed risks might be modified in order to reduce those risks. Therefore, data supporting claims of a decreased risk for delayed adverse events with novel vector types could provide the basis for reassessing the need for performing long-term follow-up observations in subjects exposed to those vectors.
Table 1. Integration Properties of Current Commonly Used Gene Therapy Vectors in Clinical Trials.
| Vector Type | Propensity to Integrate1 | Long-term Follow-up observations2 |
|---|---|---|
| Plasmid | No | No |
| PoxVirus | No | No |
| Adenovirus | No | No |
| Adeno-associated virus3 | No | No |
| Herpesvirus | No, but may undergo latency/reactivation | Yes |
| Gammaretrovirus | Yes | Yes |
| Lentivirus | Yes | Yes |
1Based on vector design (i.e., lack of any known mechanism to facilitate integration), as well as cumulative preclinical and clinical evidence suggesting that vector does not integrate or integrates only at very low frequencies.
2Specific circumstances showing persistent expression of the transgene, in the absence of integration, may be the basis for a conclusion that long-term follow-up observations are necessary to mitigate long-term risks to subjects receiving these vectors. This would depend on additional criteria, such as the transgene expressed or clinical indication, as described in the text.
3Rep-negative vectors only.
In this section, we recommend elements appropriate to the design and conduct of long-term follow-up observations.
The recommendations in this section apply to protocols for which long-term follow-up observations appear advisable. Long-term follow-up observations may be necessary to mitigate long-term risks to subjects receiving these vectors if:
In selected instances where we would generally require long-term follow-up observations, you may determine that the observations would have no scientific value based on the suitability of your clinical trial population. If you make that determination and decide not to conduct long-term follow-up, you should include in your IND the justification for your decision not to continue to observe your subject population.
The sections below provide information on criteria you may choose to use to determine the suitability of monitoring your clinical trial population to collect scientifically informative data by the performance of long-term follow-up observations. We also discuss our recommendations for the minimum duration of follow-up observations and the minimum observations to be made during long-term follow-up.
Long-term follow-up observations may have reduced utility in assessing and mitigating subject risk when the population selected for the trial has characteristics, such as short life expectancy, multiple morbidities, and exposure to other agents, that also could cause delayed adverse events. Thus, for example, long-term follow-up observations might have little impact if the subjects have widespread disease, or extensive exposure to agents with potential for delayed adverse events such as radiation or chemotherapy. In contrast, long-term follow-up observations could have greater value in assessing and mitigating the risks to subjects who have limited disease or are disease-free, and who have few co-morbidities and limited exposures to other agents with potential for delayed adverse events. In those cases where the gene therapy intervention alters life expectancy or co-morbidities, initial assessments regarding the suitability of long-term follow-up observations in a particular clinical trial may need to be reconsidered.
The duration of long-term follow-up observations should be sufficient to observe the subjects for risks that may be due to the characteristics of the product, the nature of the exposure, and the anticipated time of occurrence of delayed adverse events. The BRMAC on November 17, 2000, April 6, 2001, and October 24, 2001, discussed several different time periods for the performance of long-term follow-up observations, including a 15 year period (See Section II.B for reference). Based on the BRMAC advice, we also recommend a minimum 15 year time period for follow-up observations. However, we recognize that shorter periods of observation may be appropriate in individual trials based on supporting evidence. Elements that will influence the determination of the duration of long-term follow-up observations include the following:
Our recommendations on the nature of the follow-up observations are also based on the recommendations and discussions at the November 17, 2000, April 6, 2001, and October 24, 2001, BRMAC meetings (See Section II.B for references). As more clinical data accumulate, our recommendations regarding the duration of long-term follow-up observations may change.
It is important that the design of long-term follow-up observations be appropriate to detect potential gene therapy-related delayed adverse events in the study subjects enrolled in your clinical studies. In this document, we provide recommendations for general minimum elements for the long-term follow-up component of your study protocol.
The investigator is required to prepare and maintain adequate and accurate case histories that record all observations and other data pertinent to the investigation on each subject administered the investigational drug or employed as a control in the investigation (see 21 CFR 312.62(b)). These records would include a baseline history prior to exposure to the product in which all diseases, conditions and physical abnormalities are recorded. You are encouraged to develop a template for health care providers who are not investigators or subinvestigators (for example, the subject's physician, physician assistant, or nurse practitioner) to use in recording and reporting such observations to the investigator. Case histories should also include information from scheduled visits by a health care professional and test results for persistent vector sequences. The use of surrogate tests may be used to indicate vector persistence if direct sequence testing would require an invasive procedure for the subject.
In addition, for at least the first five years we recommend that you do the following:
For the subsequent ten years, at a minimum, we recommend that you ensure that your investigators:
Perform all long-term follow-up observations according to FDA regulations governing clinical trials (See http://www.fda.gov/oc/gcp/regulations.html). We provide additional specific recommendations and requirements for data collection and reporting of adverse events for long-term follow-up clinical observations as follows:
To increase subject compliance and improve the quality of data collection, we suggest that you encourage study subjects to monitor themselves and assist in reporting adverse events. Devices that study subjects could use to report events to the investigator include subject diaries of health-related events, informational brochures, and laminated, wallet-sized cards with investigator contact information.
The informed consent document must describe, among other things, the purposes of the research, the expected duration of the subject's participation and the procedures to be followed (21 CFR 50.25(a)(1)). Accordingly, the informed consent document must explain the purpose and duration of long-term follow-up observations, the time intervals and the locations at which you plan to request the subjects to have scheduled study visits or be contacted by other means, and details as to what those contacts will involve (21 CFR 50.25).
We provide additional informed consent recommendations for retroviral vectors in Section V.F.3 below.
The recommendations in this section apply exclusively to subjects in clinical trials who received integrating vectors, such as retroviral vectors or cells modified ex vivo by retroviral vectors. In at least two preclinical studies performed in mice, integration of genetic material from a retroviral vector into mouse cell DNA was reported to cause malignant transformation (Refs. 11 and 12). In addition, in one clinical study, three out of a total of 11 human subjects with X-linked Severe Combined Immunodeficiency (X-SCID) have developed clonal T-cell proliferation after receiving hematopoietic cells that had been modified ex vivo with a retroviral vector (Refs. 13 and 14). One of the three subjects died (Ref. 14). These leukemias were the result of the retroviral vector-derived DNA integrating into the subjects’ cellular DNA. The observation that children with X-SCID developed a malignancy after exposure to a retroviral vector (Ref. 13) has prompted us to provide additional recommendations for collection of data in studies in which subjects are exposed to integrating vectors, at this time best exemplified by retroviral vectors, including products derived from either gammaretroviruses or lentiviruses.
We recommend that you perform assays to assess the pattern of vector integration sites in relevant surrogate cells (e.g., determine whether cells carrying integrated vector sequences are polyclonal, oligoclonal, or monoclonal, with respect to vector integration patterns). We consider an assessment of the vector integration pattern to be relevant in subjects in gene therapy clinical trials involving integrating vectors if: (1) the target cells are known to have a high replicative capacity and long survival, and (2) a suitable surrogate is accessible for assay. For example, hematopoietic stem cells have a high replicative capacity and long survival; peripheral blood could serve as a surrogate for testing for vector persistence if hematopoietic stem cells were the target of your gene therapy. In those cases where peripheral blood is the surrogate, analyses on purified subsets of hematopoietic cells (e.g., lymphocytes vs. granulocytes) may be performed, if deemed appropriate to the study by you or FDA. As an alternative example, if the integrating vector is used for in vivo transduction of liver hepatocytes, you may not need to perform this analysis, since terminally differentiated hepatocytes are non-dividing cells under normal circumstances, and there is no reasonable surrogate that allows for non-invasive testing of vector persistence. Please refer to the following recommendations for developing methods and plans for performing these analyses.
If no evidence of oligo- or monoclonality is observed, we recommend that you report a summary of all analyses for the pattern of vector integration sites in narrative or tabular form in the annual report to your IND (21 CFR 312.33(b)(5)). However, if evidence of oligo- or monoclonality is observed, submit this essential information in an information amendment to the IND (21 CFR 312.31(a)). We recommend that you submit this amendment within 30 days.
Each subject in an investigation must be provided with a description of any reasonably foreseeable risks from participating in the investigation (21 CFR 50.25(a)(2)). Investigators must submit for Institutional Review Board approval the informed consent documents (21 CFR 56.109(b) and (c), 312.66). For all clinical trials in which subjects are exposed to retroviral vectors, the informed consent documents should include, in layman’s language, a complete and accurate disclosure of the development of leukemia in the children with X-SCID. We recommend that you include the following information, where applicable, in language understandable to the study subjects, in the section describing the risks associated with the study agent:
Footnotes
1This guidance does not cover the following topics:
- Inadvertent germline gene transfer. (The term “germline” is used to designate genetic material destined to be transferred to gametes). For a discussion of risks associated with inadvertent germline gene transfer for gene therapy products, we refer you to the following meeting transcripts:
- December 15-16, 1997, Recombinant DNA Advisory Committee (RAC) meeting (http://www4.od.nih.gov/oba/rac/minutes/12151697.htm),
- March 11-12, 1999, RAC meeting (http://www4.od.nih.gov/oba/rac/minutes/3-99RAC.htm), and
- November 16-17, 2000, Biological Response Modifiers Advisory Committee (BRMAC) meeting (http://www.fda.gov/cber/advisory/ctgt/ctgtmain.htm. November 17, 2000, 3664t2_b.pdf).
- Vaccines used to prevent infectious diseases even if you use products analogous to those used for gene therapy (consult the Office of Vaccines Research and Review, Center for Biologics Evaluation and Research (CBER)).
- Post-marketing or licensure requirements for performing long-term follow-up studies of subjects. The specific information needed for a licensure or post-marketing study will vary, and therefore, will be addressed with individual sponsors.
- Replication-competent non-transgene-containing viruses used as agents to mediate oncolysis. Due to the diversity of the viral agents employed, we recommend that you discuss with the Office of Cellular, Tissues, and Gene Therapies (OCTGT, CBER) the potential for risks of delayed adverse events.
- Risks due to shedding of vector to close contacts, the public, or the environment. The specifics of how and whether to address these risks in your clinical trial design should be discussed with OCTGT, CBER. For general information, see “Guidance for Industry: Environmental Assessment of Human Drug and Biologics Applications, Revision 1” dated July 1998 (http://www.fda.gov/cber/gdlns/environ.pdf).
2If you desire background information regarding prior recommendations from the BRMAC about gene therapy trials and long-term follow-up observations, we refer you to the transcripts for the November 17, 2000; April 6, 2001; and October 24, 2001, BRMAC meetings. The references can be located at http://www.fda.gov/cber/advisory/ctgt/ctgtmain.htm by searching under the year of the meeting.