|
 |
 |
|
 |
FDA Home Page | Search
FDA Site | FDA A-Z Index | Contact
FDA | FDA Centennial
![]()
| Email this Page To a Friend |
|
Many inferior food products were marketed under so-called distinctive names before passage of the Food, Drug, and Cosmetic Act of 1938. These products were heavily advertised, and were of low quality. |
By Michelle Meadows
Prompted by increasing concerns over the use of untested chemicals as food preservatives, Harvey Wiley, M.D., chief chemist of the Agriculture Department's Bureau of Chemistry, set out to investigate whether such preservatives should be used in food and which quantities were safe. Congress appropriated funds for the studies in 1902.
Wiley recruited volunteers, which the press soon dubbed the Poison Squad. The young men ate foods containing measured amounts of borax, salicylic acid, formaldehyde, and other chemical preservatives. He stopped each unconventional experiment after many of his volunteers became sick. According to anecdotal reports, none of the men was permanently harmed.
In the end, Wiley advocated that chemical preservatives should be used in food only when necessary, that the onus of safety should be on the producer of foods, and that consumers should be informed about ingredients on food labels--the basic elements of current food laws and regulations.
"The Bureau scientists watched in amazement as the public was suddenly drawn into the debates over food safety," says Suzanne White Junod, Ph.D., a historian at the Food and Drug Administration in Rockville, Md. "Wiley's studies drew widespread attention to the problem of food adulteration, and public support for passage of a federal food and drug law grew. The ‘hygienic table studies,' as Wiley preferred to refer to them, were a crucial prelude to the enactment of the Food and Drugs Act of 1906."
A century later, the FDA's Center for Food Safety and Applied Nutrition (CFSAN) still stands committed to the public, says Robert E. Brackett, Ph.D., who has served as the CFSAN's director since January 2004. "Throughout CFSAN's evolution, the dedication of the staff has remained constant, and priority-setting has always been based on doing the most good in protecting and promoting consumer health."
The CFSAN ensures the safety of about 80 percent of all food consumed in the United States, regulating everything except meat, poultry, and certain egg products, which are regulated by the U.S. Department of Agriculture (USDA). The CFSAN annually regulates 240 billion dollars' worth of domestic food; 15 billion dollars' worth of imported seafood, fresh produce, and other imported foods; and 15 billion dollars' worth of cosmetics sold across state lines. "We are proud to be a world leader in applying science-driven policies to the safety of foods and cosmetics," Brackett says.
The original Pure Food and Drugs Act was passed by Congress on June 30, 1906, and signed into law by President Theodore Roosevelt. The law, which was enforced by the Agriculture Department's Bureau of Chemistry, prohibited the introduction of misbranded and adulterated foods, drinks, and drugs in interstate commerce; prohibited the addition of color additives to conceal inferiority; and prohibited the use of "poisonous" colors in confectionary.
Under the 1906 Pure Food and Drugs Act, there was no pre-market approval system for food ingredients or drugs. The government could act only after products were on the market. On the same day that the Pure Food and Drugs Act became law, the Meat Inspection Act was passed. Historians agree that this legislation was passed amid a slump in meat sales after the publication of The Jungle, by Upton Sinclair. One chapter in the book, for example, contains shocking disclosures of unsanitary conditions in meat packing plants at the turn of the last century. The Meat Inspection Act expanded federal meat regulation to provide continuous inspection of all red meats in interstate distribution.
Other federal food legislation followed. In 1907, the Agriculture Department issued Food Inspection Decision (F.I.D.) 76, which listed seven colors approved for use in food. Subsequent F.I.D.s in the early part of the 20th century established a voluntary certification program and listed new, added colors.
In 1913, the Gould Amendment, which requires that contents be plainly marked on the outside of the food package, was added to the 1906 Pure Food and Drugs Act. A pivotal ruling on the use of a substance in food came in 1914, when the government had to show a relationship between a chemical additive and the harm it allegedly caused in humans.
The first decade after passage of the act marked a period of significant growth. "Added to initial concerns about food, and in the wake of germ theory, came increasing concerns about microorganisms in foods as a cause of disease," Junod says. "Wiley began to hire expert microbiologists. They helped transform the canning industry, the egg industry, and the refrigeration industry, helping to ensure safer food for all Americans."
But the 1906 law still had serious flaws. For one, manufacturers concerned that the new standards might shut down their businesses had inserted a so-called distinctive name proviso into the law. "This proviso allowed the marketing of foods with distinctive names that would have been otherwise illegal under the 1906 act," Junod says.
For example, a product called BRED-SPRED would have been considered adulterated or misbranded under the FDA's standards because it had no fruit. "There wasn't a single strawberry in the jar," Junod says. "It was made of coal tar, artificial pectin, artificial flavors, and grass seeds. This product and other similar products were beautifully packaged, exquisitely labeled, and heavily advertised in leading women's magazines."
Jelly had always been half sugar and half fruit. But because BRED-SPRED had a distinctive name--it didn't call itself jam or jelly--manufacturers had legal protection from misbranding. "BRED-SPRED typified the kind of inferior product that began to gain a foothold in the U.S. marketplace in the 1920s," Junod says. "Consumers had no way of knowing that such products were of low quality."
Progress was made in 1924 when the Supreme Court ruled against deceptive labeling on apple cider vinegar. According to the ruling, the 1906 Pure Food and Drugs Act prohibited information on product labels that could mislead or deceive, even if the information was technically true. But it wasn't until 1938 that the FDA received authority under a new federal food and drug statute to issue food standards in "the interest of consumers."
In 1930, the Bureau of Chemistry became the Food and Drug Administration. In 1933, the new FDA recommended a complete revision of the obsolete 1906 act. A five-year legislative battle ensued.
To show the need for a new law, the FDA put together an exhibit for Congress featuring problem products that the agency was unable to act against under the 1906 act. The collection of products included deceptive foods, dangerous cosmetic ingredients, and worthless devices and medicines.
Some of the deceptive foods in the exhibit were egg noodles that were really "plain noodles packed in yellow cellophane so that they looked like egg noodles," Junod says. "There was also chicken packed in a jar. You could see white meat on the outside, but it contained dark meat on the inside."
But it wasn't until a drug-related tragedy occurred that a new food and drug law was passed. After 107 people died from a poisonous ingredient in a product called Elixir Sulfanilamide, Congress passed the Food, Drug, and Cosmetic (FD&C) Act with new provisions in 1938.
The FD&C Act also extended regulatory control to cosmetics for the first time, in response to concerns about cosmetic safety. For example, a coal tar-based eyelash dye called Lash Lure had caused serious eye injuries, including blindness, and possibly one death.
The FD&C Act required that colors had to be listed (approved) before they could be used in foods, drugs, and cosmetics, and in addition, those made from coal tar sources had to be batch certified. The Color Certification Program, supported by user fees, continues today, with nearly 18,000,000 pounds certified for composition and purity in the FDA's laboratories in fiscal year 2005.
"More consumer-oriented than its 1906 predecessor, the 1938 Federal Food, Drug, and Cosmetic Act represents a true watershed in U.S. food policy," Junod says. "The 1938 Act eliminated the ‘distinctive name proviso' and required instead that the label of a food ‘bear its common or usual name.' The food would be illegal or misbranded if it represented itself as a standardized food unless it conformed to that standard."
The FD&C Act authorized three kinds of food standards--identity, quality, and fill of container. In 1939, the first food standards were issued for canned tomatoes, tomato purée, and tomato paste. The standards looked like a recipe of listed ingredients. The next standards were for jams and jellies. Junod says, "By 1957, standards had been set for many varieties of foods such as chocolate, flour, cereals, bakery products, milk, cheese, juices, and eggs."
In 1950, the Delaney Committee started a congressional investigation of the safety of additives that laid the foundation for the Food Additives Amendment and the Color Additive Amendments. Rep. James Delaney, D-N.Y., later submitted a change to the bill proposing the Food Additives Amendment by inserting the Delaney Clause, which prohibited the approval of any food additive shown to induce cancer in humans or animals in studies with a relevant route of exposure. Variations of the Delaney Clause were also included in the Color Additive Amendments and animal drug provisions.
Enacted in 1958, the Food Additives Amendment required manufacturers of new food additives to establish their safety to FDA's satisfaction before marketing. Food additives are substances that have no proven track record of safety and that must be approved by the FDA before they can be used. A food substance generally recognized by qualified experts as safe (GRAS) for its intended use, based on publicly available information, is excluded from the definition of food additive. Also in 1958, the FDA published the first list of GRAS substances, which contained nearly 200 substances including ascorbic acid, papain, and propylene glycol.
The Color Additive Amendments, enacted in 1960, defined "color additive" and required manufacturers to establish the safety of color additives prior to their use in coloring foods, drugs, cosmetics, and medical devices. At the time, about 200 color additives were in commercial use.
Under the 1960 Amendments, all the color additives were put on a provisional list for use on an interim basis pending evaluation by the FDA. The agency subsequently began permanently listing in the Code of Federal Regulations those color additives for which scientific data established their safety. Today, about half of the original 200 color additives are permanently listed for use in foods, drugs, cosmetics, and medical devices. Recent additions include D&C Black No. 2, approved for use in certain cosmetics, and mica-based pearlescent pigments, approved for use in contact lenses.
In 1966, the Fair Packaging and Labeling Act required products marketed on a retail basis to consumers in interstate commerce to be honestly and informatively labeled, with the FDA enforcing provisions on foods, drugs, cosmetics, and medical devices.
In 1969, President Richard Nixon convened a White House Conference on Food, Nutrition, and Health. "This important conference reinvigorated the agency's food program," Junod says. "The conference set the agency on a new path with regard to nutrition and food labeling."
In 1979, a syndrome associated with chloride deficiency was diagnosed in about 130 infants in the United States. Development of the syndrome was associated with prolonged exclusive feeding of two chloride-deficient soy formulas, Neo-Mull-Soy and Cho-Free. In 1978, the manufacturer had reformulated the products by discontinuing the addition of salt (sodium chloride). The resulting products contained an inadequate amount of chloride, an essential nutrient for infant growth and development.
To better protect infants consuming infant formula, Congress amended the FD&C Act by passing the Infant Formula Act of 1980. Under this act, the FDA requires manufacturers to follow quality control procedures, analyze each batch of formula for required nutrients, test samples for stability during the shelf life of the product, code containers identifying each batch, and maintain and make records available to FDA inspectors.
The Nutrition Labeling and Education Act (NLEA), passed in 1990, required most packaged foods to bear nutrition labeling. Food nutrition information, serving sizes, and terms such as "low fat" and "light" were standardized. The NLEA also provided for health claims.
Health claims, as defined by the NLEA, are statements supported by significant scientific agreement that show a risk reduction relationship between a food or food component and a disease or health-related condition. Examples include folic acid in breakfast cereals (reduction in risk of neural tube defects), fiber in fruits and vegetables (reduction in risk of coronary heart disease), and calcium in dairy products (reduction in risk of osteoporosis). The FDA authorized seven health claims in 1993 as part of the NLEA, and has authorized several more since then. Some examples include calcium and osteoporosis; sodium and hypertension; and fruits, vegetables, and grain products that contain fiber, particularly soluble fiber, and coronary heart disease.
In 1993, the Nutrition Facts panel, which contains basic per-serving nutritional information, was required on foods under the NLEA. "The Nutrition Labeling and Education Act was so significant because it required that food manufacturers give consumers clear and reliable information on everything from calories, fat and sodium to protein and vitamins in a format that was consistent among products and easily understood," says Janice Oliver, the CFSAN's deputy director.
The FDA's regulation of food and its education programs have kept pace with new technology, scientific advances, and the types of foods being offered to consumers, Oliver says. "The agency has continued to use state-of-the-art science and technology to ensure that consumers are given the information they need to prepare healthy, nutritious meals for themselves and their families."
The FDA established the Seafood Hazard Analysis and Critical Control Point (HACCP) Regulations in 1995. The regulations went into effect in December 1997. Based on a system developed by NASA to ensure safe food for astronauts, the HACCP is a science-based approach that requires processors to identify potential hazards that could cause food to be unsafe to eat, establish and monitor targeted critical control points to minimize risks, and keep records of the results.
Before the HACCP, industry relied on the general sanitation requirements of the FDA's Good Manufacturing Practice Regulation and testing of finished products to remain in compliance with FDA requirements. "HACCP is a more effective approach in that it is proactive rather than reactive in preventing hazards from occurring in foods," Oliver says. "The focus is on establishing a plan that identifies steps to be monitored in the process that are critical to assure the safety of foods and to correct any deviation in that step at the time it occurs."
In 1997, the Clinton Administration proposed a $43 million Food Safety Initiative to strengthen food safety practices and policies and to reduce foodborne illness in the United States. Federal agencies worked together to produce a 50-page report titled "Food Safety From Farm to Table."
The HACCP was the centerpiece of the inspections segment of the initiative. After the Seafood HACCP Regulation, the USDA developed a HACCP program for meat and poultry processing plants in 1998. The FDA established the Juice HACCP Regulations in 2001, and they went into effect in 2003 and 2004.
And in 1999, the FDA established new requirements to improve egg safety to reduce illness associated with Salmonella enteritidis. Both of these rules went into effect in 2000. Also, as part of an effort to improve the safety of imported and domestic fresh produce, the CFSAN and the USDA issued guidelines in 1998 to minimize food safety hazards for fresh fruits and vegetables. This step led to the development of the Good Agricultural Practices (GAPs) and the Good Manufacturing Practices (GMPs) for imported foods.
The Food Safety Initiative also allowed Web sites to update technology and to build a national fingerprinting database of bacterial deoxyribonucleic acid (DNA). This technology gave scientists the tools to see whether the bacteria's DNA "fingerprint" matched that of another patient or food, to identify a common source of infection. This approach speeds up the detective work to determine when an outbreak has occurred so there can be a quicker response.
The initiative also called for FoodNet (Foodborne Diseases Activity Surveillance Network) Web sites to create a more powerful network for detecting, responding to, and preventing foodborne illness. The FDA, the CDC, and the USDA support FoodNet.
The FDA is also the principal federal agency responsible for regulatory oversight of the safety and labeling of dietary supplements. The Dietary Supplement Health and Education Act (DSHEA), passed in 1994, establishes specific labeling requirements, provides a regulatory framework, and authorizes the FDA to establish current GMP regulations for dietary supplements. The DSHEA defines "dietary supplements" and "dietary ingredients" and classifies them as food.
Unlike new drugs, dietary supplements are regulated like foods. They don't have to go through review by the FDA for safety or effectiveness or be approved before they can be marketed. But manufacturers must provide pre-market notice and evidence of safety for any supplements they plan to sell that contain certain dietary ingredients that were not in the market before the DSHEA.
In 2000, the FDA published a rule on dietary supplements that defined the type of labeling claim that can be made on product labels regarding the effect of supplements on the structure or function of the body. In 2003, the FDA published a proposed rule for current GMP requirements for dietary supplements to prevent mistakes with ingredients, the presence of contaminants, mislabeling, and improper packaging.
"At the core of DSHEA enforcement efforts is our commitment to work with industry to encourage the legitimate manufacture, sale and use of supplements while enforcing the law aggressively against fraudulent product claims and other illegal practices," Brackett says.
For example, in 2004, the FDA banned dietary supplements containing ephedra, a naturally occurring substance found in plants, because of concern over their cardiovascular effects.
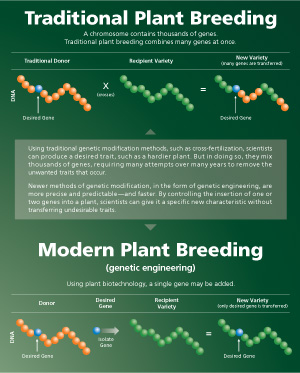
A chromosome contains thousands of genes. Traditional plant breeding combines many genes at once. Using plant biotechnology, a single gene may be added. |
Genetically engineered foods are produced from crops whose genetic makeup has been altered through a process called recombinant DNA technology, or gene splicing, to give the plant a desirable trait.
The USDA makes sure the plants do not have an adverse effect on the agricultural environment; the Environmental Protection Agency ensures that pesticides introduced into the plants are safe for human and animal consumption and for the environment; and the FDA ensures that foods made from the plants are safe to eat.
The FDA first published guidelines for genetically engineered foods in 1992. The first genetically engineered whole product was a tomato, and it went on the market in 1994. Since then, FDA has evaluated data on more than 65 other genetically engineered foods demonstrating that they are as safe as their conventional counterparts.
The CFSAN launched CAERS in 2003 to centrally help track and monitor adverse events. The CFSAN's scientists have developed rapid methods to detect microbial and viral food contaminants, and the FDA works with public and private sector partners to complete and operate two nationwide high-tech systems for rapid identification and control of outbreaks of foodborne disease.
After the terrorist attacks on Sept. 11, 2001, the FDA conducted food supply vulnerability assessments, and received additional resources for strengthening its field staff of food inspectors and lab specialists. The agency has also worked closely with the CDC and the USDA to establish the Food Emergency Response Network, a national network of laboratories ready to respond to a food security emergency.
Under the authority of the Public Health Security and Bioterrorism Preparedness and Response Act of 2002, signed by President George W. Bush in June 2002, the FDA developed four new regulations that address provisions of the law. They require food facilities to register with the FDA, to receive prior notice of imported food shipments before food arrives in the United States, and for people who receive and distribute food to keep records of their food sources and recipients. The fourth regulation establishes procedures for the FDA to detain any food for up to 30 days for which there is credible evidence or information that the food poses a threat of serious adverse health consequences or death to humans or animals.
"The possibility of food products being used as a vehicle for an attack is particularly worrisome because such an event potentially affects everyone in the United States," Brackett says. "FDA has made great strides in implementing the food-related provisions of the Bioterrorism Act. The law improves the agency's ability to prevent, prepare for, and respond to bioterrorism and other public health emergencies that affect the U.S. food supply."
To help consumers choose heart-healthy foods, the FDA announced in 2003 that food labels would have to include the amount of trans fat content. This is the first substantive change to the Nutrition Facts panel on foods since 1993, when nutrition labeling became mandatory. The requirement for listing trans fat takes effect in January 2006.
Trans fat is made when manufacturers add hydrogen to vegetable oil, a process called hydrogenation. Trans fat can be found in vegetable shortenings, some margarines, crackers, cookies, snack foods, and other foods made with or fried in partially hydrogenated oils. This process increases the shelf life and flavor stability of foods that contain these fats. Like saturated fat and dietary cholesterol, trans fat raises the risk of heart disease.
"This is an important change because the health of all consumers depends heavily on what we eat on a daily basis over a lifetime," Oliver says.
Prompted by a rising obesity rate, the FDA formed an Obesity Working Group in 2003. The working group developed an action plan to deal with the nation's obesity epidemic from the FDA's perspective. In March 2004, the group released "Calories Count: Report of the Obesity Working Group."
"Obesity will be a focus of the future because of the alarming number of adults and children in the United States who are overweight or obese, and because these are associated with increased risk of serious illness and death," Oliver says. "We will be looking at ways to both enhance the food label and improve our consumer education efforts in this area."
The FDA also worked with other Department of Health and Human Services agencies and the USDA to release the Dietary Guidelines for Americans in 2005. These guidelines are the government's science-based advice to help Americans choose diets that will meet nutrient requirements, promote health, support active lives, and reduce risks of chronic disease.
Another key area for the future is food allergens. The Food Allergen Labeling and Consumer Protection Act, passed in 2004, requires protein source labeling of any food that is or contains an ingredient that is a major food allergen. The major food allergens are peanuts; soybeans; cow's milk; eggs; fish; crustacean shellfish, such as shrimp and crabs; tree nuts; and wheat or ingredients that contain protein derived from these foods. Major food allergens account for the vast majority of food allergies in the United States.
"The labeling requirements of this act take effect beginning January 1, 2006," Brackett says. "The act requires that foods containing one or more of the major food allergens be labeled to disclose the allergen. Each year roughly 30,000 persons require emergency room treatment and 150 die because of allergic reactions to food."
The dynamic of the food supply has changed considerably in the last century, Brackett says. "Americans get food from all over the world and eat more food prepared outside the home." For this reason, the FDA encourages consumers to take proper safety precautions, such as keeping hands and surfaces clean and cooking food to proper temperatures.
"There are so many messages in the media that it can be hard for consumers to know what matters," he says. "As we face the increasing globalization of the food supply, we will keep reaching out to the public. We want to stay in touch with their interests and give them the information they need to stay healthy."
The regulatory requirements governing the sale of cosmetics are different from those that apply to most other FDA-regulated products. Under the Federal Food, Drug, and Cosmetic Act (FD&C Act), cosmetics and their ingredients, with the exception of color additives that are not coal-tar hair dyes, are not required to undergo approval by the FDA before they are marketed in products sold to the public. The FDA generally regulates these products after they have been released in the marketplace.
Manufacturers may use any ingredient or raw material, except for color additives (other than coal-tar hair dyes) and a few prohibited substances, to market a product without a government review or approval. Cosmetic firms are to ensure that the products they market are safe as used and properly labeled. Under the FD&C Act, it is prohibited to market an adulterated or misbranded cosmetic in interstate commerce. The agency can take regulatory action if it has information to support that a cosmetic is adulterated or misbranded. The FDA also works with the U.S. Customs Service and Border Protection to monitor imported cosmetics.
The FD&C Act defines cosmetics as articles intended to be applied to the human body for cleansing, beautifying, promoting attractiveness, or altering the appearance, including the components of such articles. Examples are perfumes, lipsticks, makeup, and hair dyes. Under the law, the term "cosmetic" includes both finished products and ingredients. If a product's intended use is to cure or prevent disease, or to affect the structure or function of the body, however, it may be a drug under the law. Some products are both drugs and cosmetics. For example, a dandruff treatment is a drug. A shampoo to clean and beautify hair is a cosmetic. An anti-dandruff shampoo is both a drug and a cosmetic. Products that are labeled to contain a sunscreen for sun protection are regulated as over-the-counter (OTC) drugs. Cosmetic products marketed with sun-protection claims are regulated as both drugs and cosmetics.
The FDA has taken a number of advisory and judicial actions against adulterated and misbranded cosmetics and drug products marketed as cosmetics. These include the "wrinkle remover" court cases of the 1960s and a series of Warning Letters from the 1980s to the present concerning drug claims on cosmetic products.
Another important law pertaining to cosmetics is the Fair Packaging and Labeling Act (FPLA). Under authority of the FPLA, the FDA requires an ingredient declaration on every cosmetic product offered for sale to consumers. Ingredients also must be listed in descending order of quantity.
In the 1970s, the FDA established the Voluntary Cosmetic Registration Program (VCRP), through which firms may register their establishments and file cosmetic product formulations. The VCRP provides the FDA with important information about cosmetic products and ingredients on the market in the United States.
The FDA has published a number of regulations and guidance documents specifying warning statements required for certain cosmetics, as well as requirements for the way in which required information must be displayed on cosmetic labels. Most recently, a "Sunburn Alert" was published for cosmetic products that contain alpha hydroxy acids, ingredients that may be found in anti-wrinkle products.
Scientists at the CFSAN's Office of Cosmetics and Colors (OCAC) continue to keep abreast of new ingredients and safety concerns. For example, current research at the OCAC includes cosmetic applications of nanotechnology and its implications for cosmetic safety.
The FDA's Center for Food Safety and Applied Nutrition
www.cfsan.fda.gov/
![]()