


Report to Congress
Reports on Postmarketing Studies
[FDAMA 130]
- INTRODUCTION
The sponsor of a drug that has entered into an agreement with FDA to conduct a postmarketing study must submit a report annually on the progress of the study and the reasons, if any, for failing to complete the study.
FDA must publish annually in the Federal Register a report that provides information on the status of postmarketing studies that sponsors have agreed to conduct and for which annual reports have been submitted.
FDA must prepare a report to Congress by October 1, 2001 that includes:
A summary of annual status reports submitted;
An evaluation of sponsor performance in fulfilling agreements with respect to conducting postmarketing studies;
The timeliness of FDA's review of postmarketing studies; and
Any legislative recommendations respecting postmarketing studies.
FDA must consider information pertaining to the annual report as public information to the extent that the information is necessary to identify the sponsor; and to establish the status of the study and the reasons, if any, for failure to carry out the study.
BACKGROUND
The Need for Postmarketing Studies
- Accelerated approval clinical benefit studies. In December 1992, FDA published new regulations [21 CFR 314, subpart H and 601 subpart E] to establish a process for FDA to grant marketing approval under an accelerated review process for products that treat serious and life-threatening illnesses and that provide meaningful therapeutic benefit over existing therapies. Congress incorporated this approach into law in Section 112 of FDAMA (Section 506 of the act; 21 U.S.C. 356).
Deferred pediatric studies. FDA implemented regulations in April, 1999 requiring applicants of drugs and biologics that are used in, or have the potential to be used in, pediatric populations to assess the safety and effectiveness of those products in all relevant pediatric populations [21 CFR 314.55 and 601.27]. This rule applies to both drugs under review and certain already marketed drugs. For drugs under review, FDA may permit these studies to be deferred until after the drug is approved in adults. When deferred, these assessments are performed as postmarketing studies. If these studies are not completed and reported, FDA may deem the product to be misbranded.
Experience with Postmarketing Studies
OIG Report
- formal standards or procedures for monitoring or for establishing whether a postmarketing commitment has been met; and
- an effective management tool for tracking postmarketing commitments.
- establish standards, procedures, or guidelines for individuals responsible for carrying out monitoring and tracking objectives; and
- establish accountability for monitoring, tracking, and bringing commitments to closure.
FDAMA IMPLEMENTATION of FDAMA SECTION 130
Annual Report Requirements.
On December 1, 1999, FDA published for comment a new proposed rule requiring annual postmarketing study progress reports. The final rule issued on October 30, 2000 and after the effective date was delayed once for 60 days, became effective April 30, 2001. The Agency began receiving annual progress reports under the new regulation [21 CFR 314.81(b)(2)(vii) and 601.70] in May 2001. All applicants with postmarketing study commitments must submit a progress report by October 30, 2001.
Modify postmarketing reporting for drugs. Annual report requirements for drugs [21 CFR 314.81(b)(2)] have long required a summary statement on the status of postmarketing studies. Information submitted in NDA annual reports has not always allowed FDA to determine how a study was progressing. Modifications to the regulation now require specific information to allow a determination of the progress of studies.
Create a reporting requirement for biologics. Although many biologics license applicants have been requested to perform postmarketing studies, there has been no regulatory requirement for reporting the status of such studies. New regulations [21 CFR 601.70] now require annual reporting on the status of postmarketing studies for biologics approved under the PHS Act;
Define scope of reporting requirement. New regulations [21 CFR 314.81(b)(2)(vii) and 601.70] limit the reporting requirements to studies addressing those most pertinent to improved use of a drug or biologic [i.e., clinical efficacy, clinical safety, clinical pharmacology or non-clinical toxicology].
Establish the content and format of annual progress reports. New regulations [21 CFR 314.81(b)(2)(vii) and 601.70] specify a format and describe the content of annual reports on the progress of postmarketing studies. To minimize the reporting burden on applicants and the review burden on the Agency, these format and content regulations require specific, but minimal information necessary to identify the applicant, the product, and the postmarketing study; the study status; and a brief explanation of the study status.
Clarifies public information. New regulations [314.81(b)(2)(vii) and 601.70] clarify that certain information necessary to identify the applicant, the study and its status will be considered public information.
- Improved Management Tools. The Agency has made several procedural improvements and has developed data tracking systems to improve its monitoring and processing of annual status reports and study final reports.
Administrative Procedures: CDER is currently updating revising staff operating procedures to clarify how postmarketing commitments and the annual status reports will be processed, reviewed, and archived. The Center for Biologics Evaluation and Research (CBER) completed this process in May 2001 and issued a new standard operating procedure [SOPP 8413: Postmarketing Commitment Annual Reports, Final Reports, and Related Submissions - Administrative Handling, Review, and CBER Reporting].
Data Tracking: Both CBER and CDER have developed new data systems to more efficiently capture the existence of postmarketing commitments; the submission of annual study progress reports, the submission of study final reports, and final review determinations. The databases were implemented in July, 2000 at CBER and July 2001 at CDER. The databases will be updated as submissions are received and reviews completed. The databases will be used to provide information to a public FDA Web site on postmarketing studies.
- Information to the Public. FDA has plans for providing information on postmarketing studies through various mechanisms including reporting via FDA's Web site and the Federal Register.
Web site: FDA plans to place on its public Web site a report on all postmarketing status reports received. This will allow easy access to public information from study status reports. This information is designed to make patients and their families aware of postmarketing studies that are being conducted and to allow them to follow the progress applicants are making in completing those studies. The Agency also intends to identify on the Web site those studies for which the study commitment has been fulfilled. As planned, the Web site would include the following information:
Name of Applicant;
Product Name/Proprietary Name(s);
Route of Administration/Dosage Form/Strength;NDA, ANDA, or BLA Number;
Date of US Approval;
Postmarketing Study Commitment:
Annual Report Due Date;
Date Report Received.
Federal Register Report: In October, 2002 FDA will initiate an annual Federal Register report on industry performance on performing postmarketing studies and in meeting postmarketing commitments. The target date of October 2002 was chosen because it allows applicants a full year of reporting and provides a sufficient basis for the Agency to determine how the industry is performing with respect to completing postmarketing commitments. The Agency plans to provide the following information in its Federal Register report:
The number of applicants with outstanding postmarketing study commitments;
The number of outstanding postmarketing study commitments;
The number of outstanding study commitments for which no reports were submitted to FDA;
The number of concluded studies that
satisfied an applicant's commitment;
failed to satisfy an applicant's commitment;
FDA deemed no longer feasible or needed.
SPECIFIC ISSUES TO BE REPORTED UNDER SECTION 506B
a summary of the reports submitted under section 506B of the Act; and
an evaluation of:
the performance of sponsors completing postmarketing studies;
the timeliness of the Secretary's review of the postmarketing studies; and
any legislative recommendations respecting the postmarketing studies.
Summary of Reports Submitted under Section 506B
- Pending: The study has not yet begun.
- Ongoing: The study is proceeding according to, or is ahead of the applicant's original schedule.
- Delayed: The study is proceeding but is behind the applicant's original schedule.
- Terminated: The study was ended before completion.
- Submitted: The study has been concluded or terminated and the applicant has submitted a final study report to FDA.
Evaluation of Applicant Performance in Fulfilling Agreements to Conduct Postmarketing Studies:
Timeliness of FDA's Review of Postmarketing Studies
- Study final reports submitted as supplemental applications: review within established PDUFA timeframes;
- Study final reports (no supplement): review within 12 months.
Legislative Recommendations Respecting Postmarketing Studies
Failure to complete studies under accelerated approval may result in a withdrawal of approval [withdrawing the drug from the market] or modification to labeling claims.
Failure to complete deferred pediatric studies and to submit pediatric labeling may cause the product to be misbranded. FDA may initiate seizure or injunction actions.
CONCLUSIONS
FDAMA Section 130 has provided FDA with tools to monitor the conduct of postmarketing studies important to improved drug and biologic use and safety.
FDA will use these tools to monitor industry compliance in conducting postmarketing studies and in ensuring timely FDA review of information from completed studies;
Useful information concerning postmarketing studies is being made available to the public.
FDA needs resource support to optimize its efforts in surveillance of applicant compliance with postmarketing commitments and to conduct timely review of information resulting from completed postmarketing studies.
FDA will inform Congress if regulated industry fails to comply with postmarketing commitments and will seek authority for additional penalties.
APPENDIX
In 1997, Congress passed the Food and Drug Administration Modernization Act (FDAMA) amending the Federal Food, Drug, and Cosmetic Act (the Act). In addition to amending numerous existing sections of the Act, Congress added several new provisions.
New section 506B (21 U.S.C. 356b) provides additional authority for monitoring the progress of postmarketing studies that drug and biologic applicants have agreed to conduct. Congress enacted this section in response to concerns expressed by the Food and Drug Administration and the public about the timeliness of completing postmarketing studies and about the need to update drug labeling with information obtained from such studies. The new section requires drug sponsors and FDA to do the following:
To implement section 506B, FDA has defined the content and format of annual progress reports for postmarketing studies. The Agency has also made modifications to a number of internal operating procedures and programs to more efficiently track and monitor the status of postmarketing studies. This report first briefly discusses the need for postmarketing studies, provides a discussion of background activities related to postmarketing studies, and describes steps taken by the Agency to implement section 506B. The report then examines the specific issues mandated by Congress.
Before drug companies can market new prescription medicines (drugs and biologics) in the United States, they must submit applications to FDA and receive approval of the applications. Normally, applicants test new drugs and biologics in three phases of studies before applying for marketing approval. These premarket studies gradually introduce experimental products to larger numbers of patients to determine the product's safety and efficacy under controlled conditions.
Before or after granting marketing approval, FDA may ask the manufacturer to conduct a "phase 4" or "postmarketing study." This request is made if FDA concludes that additional information, while not essential for approval, is important in improving the prescribing and use of the product; product quality; or consistency in product manufacturing. Postmarketing studies may confirm existing data, raise or answer questions, or provide new data.
Mandatory postmarketing studies have been incorporated into the regulation of two types of products: 1) Fast track products, approved on an accelerated basis and 2) products for which safe use in children needs to be determined or more clearly defined.
Under the accelerated approval process, FDA may approve products based on a surrogate marker or other clinical effect that is reasonably likely to predict clinical benefit, provided that the applicant conducts postmarketing studies to verify and describe the clinical benefit when there is uncertainty about the relation between the data submitted and clinical benefit or ultimate outcome. The accelerated approval process allows the product to enter the market sooner but with less complete clinical efficacy information than the standard review process requires. When using the accelerated approval process, FDA may require postmarketing studies to gather complete efficacy information and can withdraw marketing approval if the studies are not completed with due diligence or if the studies fail to verify clinical benefit of the product.
In addition, FDA has authority to order an applicant to conduct a study if the information is necessary in order to facilitate a determination whether grounds exist for revocation of approval (21 U.S.C. 355(k)). Moreover, applicants frequently recognize the importance of postmarketing studies in the context of a particular application, and, at FDA's request, commit to the Agency to conduct them. A company's agreement to conduct a study is called a postmarketing commitment. In this report we use the term commitment when discussing both agreed-upon and required postmarketing studies for drugs and biologics.
For many years, regulations for New Drug Applications (NDAs) have required that information on the status of postmarketing studies be included in NDA annual reports [21 CFR 314.81]. For biologics licensed under the Public Health Service (PHS) Act, there has been no comparable requirement for reporting the status of postmarketing studies.
Postmarketing studies are essential in assessing optimal use of drugs and biologics. Results from such studies have made significant changes to how products are used. Listed below are a few examples of improvement to health care through better treatment regimens or decreased toxicity:
Interferon alfa-2b [INTRON A] This biologic was originally approved to treat chronic hepatitis C with a 6-month treatment regimen. The applicant agreed to conduct postmarketing studies to determine the optimum duration of therapy by studying longer treatment times. Results showed that treatment for at least 12 months provided a significantly higher rate of efficacy, doubling the number of patients obtaining benefit from the product.
Abciximab [ReoPro] This biologic was approved based on its efficacy in preventing cardiac ischemic complications in patients undergoing coronary angioplasty. A major complication with use of this drug was a high rate of bleeding events. Postmarketing studies investigated ways to reduce bleeding and found that modifications to the way the product was given as well as adjusting drugs used in conjunction with ReoPro could maintain the beneficial effects while significantly reducing the number and severity of bleeding events.
Fluvoxamine [LUVOX Tablets] This drug was originally approved for the treatment of obsessions and compulsions in patients with Obsessive Compulsive Disorder (OCD). After approval in adults, effectiveness in children was explored and Luvox was approved to treat children and adolescents, ages 8-17 at a substantially lower dose. Exploratory analyses performed during review of the indication in children showed effect in the 8-11 age group and essentially no effect in the 12-17 age group. Because of this lack of effect in teens, the applicant committed to conduct a postmarketing pharmacokinetic study. This study showed a 2-3 fold higher drug level in children compared to teens. This led to labeling changes that now reflect the increased doses required in teenage patients.
Itraconazole [Sporanox] Janssen submitted the results of a voluntary study that showed heart effects in patients treated with Sporanox. FDA discussed its concerns about the results of the study with representatives from Janssen and requested that the firm conduct an additional postmarketing study. Following the completion of the study, Janssen concluded the heart effects could be of concern. The Office of Postmarketing Drug Risk Assessment of FDA's Center for Drug Evaluation and Research (CDER) also conducted an epidemiological review and discovered cases of heart failure in patients on Sporanox therapy. FDA requested Janssen update the Sporanox labeling to include the cardiac risk. Janssen revised the label by adding a new boxed warning about the heart effects and issued a "Dear Health Care Provider" letter to physicians and health care providers. In addition, FDA issued a Public Health Advisory and Talk Paper to inform consumers of the recently identified risk.
FDA has a long history of requesting that postmarketing studies be performed when there is reason to believe additional data from such studies will improve how a product is used. The graphs below provide some historical perspective on FDA's experience with postmarketing commitments.
Figure 1 presents a bar graph of the number of postmarketing commitments associated with Biologic License Applications (BLAs) approved between 1991-2000. During this 10 year period, 163 BLAs were approved, 79 of which had at least one postmarketing commitment. These BLAs, combined with supplemental applications received during this period, generated a total of 927 postmarketing commitments. Of this number, 193 commitments addressed clinical safety and efficacy studies and would be subject to status reporting under FDAMA 130 implementation.

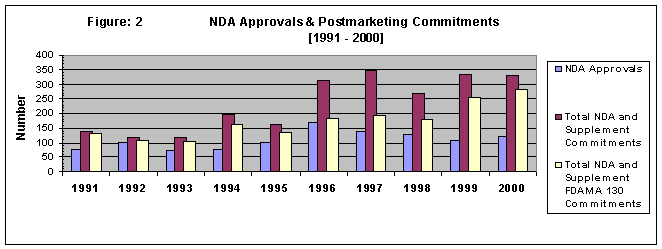
Figure 2 presents a bar graph of the number of postmarketing commitments associated with New Drug Applications (NDAs) approved between 1991-2000. During this period, 1,090 NDAs were approved. These NDAs plus supplemental applications generated a total of 2,328 postmarketing commitments. Of these, 1,737 commitments addressed clinical safety and efficacy and would be subject to status reporting under FDAMA 130 implementation.
The Office of the Inspector General (OIG) issued a report on its investigation of postmarketing studies for prescription drugs on May 1, 1996. The investigation focused on new molecular drug entities (NME) approved by the CDER between 1987 and 1993. Agency data showed that 53% of the NMEs had one or more postmarketing commitments. Although postmarketing studies were a common means of generating data to further delineate the Agency's knowledge concerning use of the drug, the report identified a number of deficiencies that negatively affected completion of commitments and/or use of the data the studies generated. Included in the OIG list of concerns was the observation that the Agency lacked:
The report observed that because of these deficiencies, the status of many commitments was unknown and the time taken by the Agency to review some study final reports ranged between a few months to several years.
In its recommendations to FDA, the OIG acknowledged that the Agency's ability to monitor postmarketing studies was hampered by 1) its priority in reviewing premarketing studies; 2) limited resources, a situation which was worsened by shorter review times for premarketing submissions as required under the Prescription Drug User Fee Act of 1992; and 3) inability to enforce compliance in performing postmarketing studies. The OIG recommended that FDA:
Following enactment of FDAMA, FDA initiated a number of steps to implement the provisions of Section 130 on postmarketing studies.
These new regulations [21 CFR 314.81(b)(2)(vii) and 601.70] accomplish the following:
The Agency has also published a draft guidance to facilitate the implementation of the annual reporting requirements:
Draft Guidance for Industry: Reports on the Status of Postmarketing Studies-Implementation of Section 130 of the Food and Drug Administration Modernization Act of 1997 (April, 2001). See attached appendix.
The public comment period on this guidance closed June 4, 2001. The Agency is evaluating these comments before finalizing the guidance.
The Centers have designated individuals to be responsible for monitoring the submission of reports. Target review timelines have been established for annual status reports and for the review of study final reports. These timelines will be tracked and monitored. Procedures have also been established to direct how FDA staff will communicate with applicants when the review of study final reports has been completed.
-
Commitment Date
Type of Study
Commitment Description
Projected Study Completion Date
Current Status of Study
Explanation of Study Status
Section 506B requires that the Secretary of Health and Human Services (the Secretary) submit a report to Congress containing:
As of February 8, 2002, status reports have been received for 701 drug products and for 15 biologic products. These reports are currently under review in FDA. The progress of each study reported will be compared to the applicant's schedule for conducting the study. The study will then be categorized and reported under one of the following categories:
CBER and CDER analyzed their data as of February 8, 2002 on postmarketing commitments addressing clinical safety, clinical efficacy, clinical pharmacology and non-clinical toxicology.
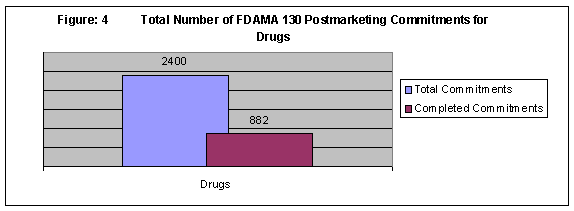
Figures 3 and 4 are bar graphs representing the total number of postmarketing commitments (completed and not-completed) for biologics and drug products requiring status reporting under FDAMA 130, respectively. The numbers of commitments include those associated with changes to approved applications (supplements) as well as applications (BLA, NDA). For biologics (BLA), 44 of 301 commitments have been categorized as completed. Of the 2400 drug (NDA) commitments, 882 have been categorized as completed.

Both CDER and CBER have used a system for listing all postmarketing study commitments in databases for several years. Although the databases contain an accurate listing of commitments, they have not been consistently used to track and monitor the progress of performance in completing postmarketing commitments. In the past, results from postmarketing studies have been submitted in various formats. For example, results from these studies have sometimes supported new indications for product use and/or otherwise altered the approved labeling. Some studies have provided information resulting in changes in the product manufacturing processes. In these cases, final study reports are submitted as supplements to drug applications. Since these studies are then reviewed and acted upon as application supplements, the relationship to postmarketing study commitment results has not always been identified and captured in the postmarketing commitment databases.
The recent changes to annual reporting requirements, Agency procedures, and updates to the database system (as described above) will allow FDA to capture post-marketing study commitment status and completion information as well as the initial commitment information.
As described for number "2" above, the previous system for tracking postmarketing studies was essentially a listing of postmarketing studies, with no system in place for capturing review and decisions regarding these studies. As also described, the new system and procedures put in place along with the changes to annual reporting requirements will provide the necessary information to report on both the evaluation of applicants fulfilling postmarketing commitments and FDA's performance regarding review of postmarketing studies.
Both CBER and CDER have set goals for review and action regarding final reports on postmarketing studies. These goals are as follows:
We will continue to report on the performance of applicants and the Agency via the annual Federal Register report planned to first publish October, 2002 (as described above).
Penalties for enforcement
FDA currently has authority outlined in regulations to take legal action against applicants who fail to complete postmarketing studies performed under accelerated approval provisions [21 CFR 314, subpart H and 601 subpart E] and deferred pediatric studies [21 CFR 314.55 and 601.27].
The above enforcement authorities have worked well when clinical benefit has not been adequately demonstrated or when patient safety has been at risk. It is our intent to diligently monitor industry and FDA performance concerning the conduct of postmarketing studies. If non-compliance is a problem, FDA intends to provide information to Congress in support of a request for additional legal authorities.
Draft Guidance for Industry: Reports on the Status of Postmarketing Studies-Implementation of Section 130 of the Food and Drug Administration Modernization Act of 1997 (Draft April, 2001) (PDF), (Text)