|
 |

|
Print  Download Reader
Download Reader 
|
|
|
Print Download Reader
Testimony
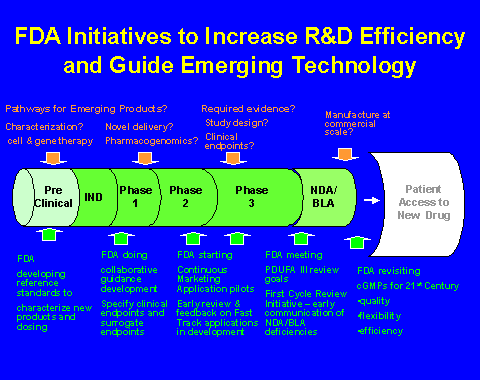
Wednesday, September 7, 2005 Introduction I particularly am pleased to be with you today, during Gynecologic Oncology Awareness Month and Ovarian Cancer Awareness Month, to discuss the topics of prevention, early detection and treatment of gynecologic cancers. My testimony will focus more on the treatment of these cancers since it is the Mission of FDA in this area to promote and protect the public health by assuring the safety, efficacy and security of human and veterinary drugs, biological products and medical devices by helping to speed innovations that make medicines more effective, safer and more affordable and to help the public obtain the accurate, science-based information they need to use these medicines to improve their health. I also will share with you what our Agency is doing to accelerate the delivery of innovative cancer treatments to meet the needs of cancer patients and their families. Further, I will discuss the Agency's interaction with other government agencies, drug sponsors and the medical professional community in an effort to streamline and accelerate the overall development of diagnostic, preventive and therapeutic interventions for cancer, as well as FDA's Critical Path Initiative. In my remarks, I will use the term "drug" to refer to both traditional small molecules and to therapeutic biological products. Recent Consolidation of Oncology Review Functions at FDA The Office also is to develop and lead a comprehensive Oncology Program to facilitate coordination of oncology activities across all Centers of FDA, and ensure ongoing outreach and collaboration between FDA, the National Cancer Institute (NCI) and other cancer-related organizations within and outside of the government. This cross cutting Oncology Program is to facilitate cross Agency expert consultation, provide a forum to discuss and develop regulatory policy and standards and serve as a focal point for Agency interaction and collaboration with oncology professional societies, NCI and other important stakeholders. The program also is to coordinate cross cutting training and oncology education programs. The Office expects to improve the consistency of review and policy toward oncology drugs and bring together a critical mass of oncologists who will help guide the development of new therapies. Although many details of this new structure are still evolving, I am extremely pleased to be working with the many talented and dedicated scientists who comprise the Office, in order to realize FDA's vision for it. Clinical Trials � The Phases of Clinical Trials Most clinical trials are carried out in consecutive steps called phases. Each phase is designed to gather different types of information. Patients may be eligible to participate in studies in different phases, depending on their general condition, the type and stage of their cancer, and what therapy, if any, they already have had. Patients are seen regularly by the investigators during the study to determine the effect of the treatment, and treatment is stopped if side effects become too severe. The purpose of a Phase 1 clinical trial is to find the best way to administer a new treatment and learn how much of it can be given safely. In a Phase 1 study, a new treatment is given to a small number of patients. For a new drug, the study starts by giving a low dose of the drug and, if necessary as preliminary findings of the trial suggest, the dose may then be adjusted as new patients enter the trial. Phase 2 studies are designed to find out whether a treatment has the intended effect. In the context of cancer therapy, Phase 2 studies are designed to study whether the treatment actually damages cancer cells or slows their growth in people. Usually groups of 20 to 50 patients with one type of cancer receive an investigational treatment in Phase 2 studies. For example, patients with breast cancer who no longer respond to standard therapy may choose to be treated in a Phase 2 study. Patients are observed closely for anti-cancer effect by repeated measurement of tumor size to see whether tumors have shrunk since the beginning of the trial. Phase 3 studies usually compare a new treatment that appeared to have an effect in the small Phase 2 studies with standard (generally accepted) therapy, or compare the combination of the new therapy and standard therapy to standard therapy alone. Phase 3 trials require larger numbers of patients; some trials enroll hundreds or even thousands of patients. Patients usually are randomized (assigned by chance) to the treatments being studied. The group that receives the standard treatment is called the "control" group. The researchers expect that a certain number of these patients will be helped by the treatment. Phase 4 trials may be conducted after a drug has been approved. Companies often, for example, carry out studies of new drugs in patients with different tumors or with different stages of disease. FDA also may request, and the sponsor may agree to conduct, other post-marketing studies to provide additional data to improve the safe and effective use of the drug. Clinical Trials for Cancer Therapy In addition to FDA review of a protocol submitted to an IND the protocol also is subject to oversight by a local Institutional Review Board (IRB). An IRB is a panel of scientists and non-scientists that oversees clinical research, and approves the initiation of the protocol at their respective institution. Experienced clinical investigators give the drug to a small number of cancer patients who have no other available therapy. These phase 1 studies assess the most common acute adverse effects and examine the amount of drug that patients can take safely without unacceptable side effects. Initial clinical studies also begin to clarify what happens to a drug in the human body, how it is changed (metabolized), how much of it (or a metabolite) gets into the blood and various organs, how long it stays in the body, and how the body gets rid of the drug and its effects. If Phase 1 studies do not reveal major problems, such as unacceptable toxicity, the next step is to conduct a clinical study in which the drug is given to patients who have medical conditions that may benefit from the potential cancer drugs. Several different types of cancers often are explored in these Phase 2 studies. Researchers then assess whether the drug has a favorable effect on the condition. Testing experimental drugs in people inevitably presents ethical questions. A general principle, agreed on internationally, is that patients in a study must not be denied known effective treatment that prevents death or serious injury. In cancer trials, patients are never denied such treatment. FDA recommends that anyone interested in participating in a clinical trial discuss the idea with his or her physician. Doctors may be able to provide information on investigational drugs that might be of benefit to their patients and of clinical trials involving these drugs. Patients can obtain detailed information from a variety of sources, including drug sponsors, FDA (if the information is public), and NIH. In fact, industry-sponsored trials are required statutorily to be listed on www.clinicaltrials.gov. Clinical trials are carried out at major medical research centers, at NIH, and even in doctors' offices. Although they may involve hospitalized patients, many clinical trials can be conducted on an outpatient basis, with participants more or less going about their normal activities. The center or institution where a study is to be carried out may run newspaper advertisements recruiting potential participants for clinical studies that tell readers where to call or write for further information. These aspects and other implications of taking part in a clinical trial must be explained fully in advance by the people conducting the trial, and patients must agree to the conditions before they can participate. The hope of personally benefiting from a new drug or the desire to take part in research that might one day benefit millions is what makes people volunteer for clinical trials. It should not prevent them, however, from finding out all they can about being a part of the process. They also must understand that new treatments, although promising, may prove ineffective or harmful. Expediting Approval of Cancer Therapies FDA programs codified in FDAMA include:
Fast-track designation for a clinical development program can occur at any time of the development process. It is initiated by the sponsor's request for designation and can be granted for any development program (as projected by the sponsor) that is intended to demonstrate that its drug/biologic will affect a serious or life-threatening disease or condition. This may be an improvement over existing therapy or treatment where no alternative therapy exists. Recently two exploratory pilot programs were instituted to build on the current practice of interaction between FDA and applicants during drug development and application review.
It is important to note that FDAMA did not alter FDA's effectiveness standard, except by giving explicit authority to the Agency to rely on data from a single, adequate and well-controlled clinical investigation and confirmatory evidence as support for approval in certain cases. Even for drugs intended for serious and fatal illnesses, there must be substantial evidence that the drug will have the effect it purports to have. As noted, however, the law recognizes that the nature of the effect that needs to be demonstrated might vary depending on the urgency and clinical need. Planned Workshop on Ovarian Endpoints In late 2002, FDA embarked on a project to evaluate potential endpoints for cancer drug approval. Endpoints have been examined for the most common cancers: lung, colon, and prostate cancer. For each cancer, FDA held public workshops to identify important issues, and these issues were later discussed in meetings of the Oncologic Drugs Advisory Committee (ODAC). Subsequently, guidance documents will be published describing FDA's current thinking on endpoints for cancer drug approval. In June 2005, FDA co-sponsored a workshop with the American Society of Hematologists (ASH) to explore endpoints in acute leukemias. Expanded Access to Investigational New Drug Products List of Drugs Approved for Treatment of Ovarian Cancer FDA Ooffice of Special Health Issues Patients usually call to obtain information about unapproved treatments currently being researched. Once our staff explains that FDA cannot disclose certain confidential information about drugs or devices that are not yet approved, we direct callers to listings of clinical trials where they can locate a trial for which they might be eligible. We are able to talk with patients about any treatment that appears in a public access database, such as the ClinicalTrials.gov database operated by the National Library of Medicine or NCI's database at http://cancertrials.nci.nih.gov. Our staff is working actively with the National Library of Medicine and the pharmaceutical industry to include more clinical trials in the ClinicalTrials.gov database. If a patient does not have a computer, a patient can access the NCI's clinical trials listing by calling 1-800-4-CANCER. An information specialist will search the database and send the trials information to the patient within 3 days. Our goals in serving patients with life-threatening diseases and their family members are straightforward:
FDA/Sponsor Interaction During Clinical Trials and The Drug Review Process Tht NCI/FDA Interagency Oncology Task Force (IOTF) Since its formation, the members of IOTF collaboratively have undertaken an analysis of the overall development and review process for new oncology drugs and devices and identified several specific initiatives that are directed toward optimizing drug and device development. NCI is working to specifically gather and synthesize the scientific support needed by FDA to address specific regulatory issues. FDA is working cooperatively with NCI to address important scientific issues including:
The IOTF is meeting regularly and actively addressing issues that can ultimately speed the development of new advanced interventions for cancer. The IOTF subcommittees are currently developing resource materials that will assist investigators in preparing the data needed for FDA's regulatory process. FDA has already responded with guidance documents (such as a recent guidance on pharmacogenomics) and process changes. FDA's Critical Path Initiative Today's revolution in biomedical science has raised new hope for the prevention, treatment, and cure of serious illnesses. However, there is growing concern that many of the new basic science discoveries made in recent years may not yield quickly more effective, affordable, and safe medical products for patients. This is because the current medical product development path is becoming increasingly challenging, inefficient, and costly. During the last several years, the number of new drug and biologic applications submitted to FDA has declined significantly; the number of innovative medical device applications also has decreased. In contrast, the costs of product development have soared over the last decade. Because of rising costs, innovators often concentrate their efforts on products with potentially high market return. Emerging contenders for resources include the development of products targeted for important public health needs (e.g., counter terrorism), less common diseases, prevalent third world diseases, prevention indications, or individualized therapy is becoming increasingly challenging. In fact, with rising health care costs, there now is concern about how the nation can continue to pay even for existing therapies. If the costs and difficulties of medical product development continue to grow, innovation will continue to stagnate or decline and the biomedical revolution may not deliver on its promise of better health. Attachment C to this testimony demonstrates this for drugs and biologics through 2002. A problem, in FDA's view, is that the applied sciences needed for medical product development have not kept pace with the tremendous advances in the basic sciences. The new science is not being used to guide the technology development process in the same way that it is accelerating the technology discovery process. For medical technology, performance is measured in terms of product safety and effectiveness. Not enough applied scientific work has been done to create new tools to get fundamentally better answers about how the safety and effectiveness of new products can be demonstrated, in faster time frames, with more certainty, and at lower costs. In many cases, developers have no choice but to use the tools and concepts of the last century to assess this century's treatment candidates. As a result, the vast majority of investigational products that enter clinical trials fail. Often, product development programs must be abandoned after extensive investment of time and resources. This high failure rate drives up costs, and developers are forced to use the profits from a decreasing number of successful products to subsidize a growing number of expensive failures. Finally, the path to market, even for successful candidates, is long, costly, and inefficient, due in large part to the current reliance on suboptimal assessment methods. A new product development toolkit -- containing powerful new scientific and technical methods such as animal or computer-based predictive models, biomarkers for safety and effectiveness, and new clinical evaluation techniques -- is needed urgently to improve predictability and efficiency along the critical path from laboratory concept to commercial product. Superior product development science is needed to address these challenges -- to ensure that basic discoveries turn into new and better medical treatments. More efforts need to be directed at creating better tools for developing medical technologies. Finally, we need a knowledge base built not just on ideas from biomedical research, but also on reliable insights into the pathway to patients. FDA is planning and beginning an initiative that will identify and prioritize (1) the most pressing development problems and (2) the areas that provide the greatest opportunities for rapid improvement and public health benefits. This will be done for all three dimensions along the critical path -- safety assessment, evaluation of medical utility, and product industrialization. It is critical that we enlist all relevant stakeholders in this effort. We are in the final stages of developing a Critical Path Opportunity List, based on the input and ideas contributed both by external stakeholders and FDA reviewers. Concurrently, FDA has refocused its internal efforts to ensure that we are working on the most important problems and intensified our support of key projects. We are working closely with NCI under the IOTF on proposals to advance the science of cancer drug development. Through scientific research focused on these challenges, we can improve the process for getting new and better treatments to patients. Directing research not only to new medical breakthroughs, but also to breakthrough tools for developing new treatments, is an essential step in providing patients with more timely, affordable, and predictable access to new therapies. We are confident that, with effective collaboration between government, academia, and the private sector, these goals can be achieved. Conclusion Thank you for this opportunity to testify. I will be happy to answer any questions the Subcommittee might have. FDA Approved Treatments for Gynecological Cancers
Last Revised: September 14, 2005 |
|||||||||||||||||||||||||||||||||||||||||||