|
About CDER - Report |
U.S. Food and Drug Administration • Center for Drug Evaluation and Research |
|
About CDER - Report |
|
|
CDER Report to the Nation: 2002 Adobe Acrobat version of this document International ActivitiesIndex
MissionWe participate through appropriate processes with representatives of other countries to reduce the burden of regulation, harmonize regulatory requirements and achieve appropriate reciprocal arrangements. Highlights from 2002 include:
International Conference on HarmonizationHarmonization-making the drug regulatory processes more efficient and uniform-is an issue that is important not only to Americans, but to drug regulatory agencies and pharmaceutical companies throughout the world. The International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use has worked to bring together government regulators and drug industry experts from innovator trade associations in the European Union, Japan and the United States. We are leading the FDA's collaboration with the ICH. This work will help make new drugs available with minimum delays not only to American consumers but also to patients in other parts of the world. The drug regulatory systems in all three regions share the same fundamental concerns for the safety, efficacy and quality of drug products. Before ICH, many time-consuming and expensive technical tests had to be repeated in all three regions. The ICH goal is to minimize unnecessary duplicate testing during the research and development of new drugs. The ICH process results in guidance documents that create consistency in the requirements for product registration.
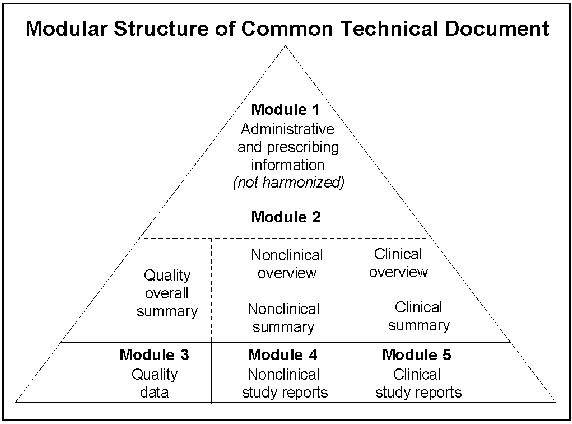
Common Technical DocumentThe ICH Common Technical Document allows data in the same format to be submitted to drug review authorities in all three ICH regions. Last year, we completed work on making the document suitable for electronic submission. ICH guidance documentsLast year we published seven ICH guidance documents. As of April 30, 2003, we had published 45 final documents and eight drafts. Harmonization initiative in the AmericasWe are working with the Pan American Health Organization to promote regulatory harmonization within the Americas. PAHO is part of the United Nations system, serving as the World Health Organization's regional office for the Americas. The initiative, called the Pan American Network for Drug Regulatory Harmonization or PANDRH, will search for common ground on various topics in a prioritized work plan. We are the lead for two topics of high priority-good manufacturing practices and bioequivalence. We are working with the countries of Latin America to provide training on these two important issues. Training to the same standards should help lead to harmonization. Other urgent issues are good clinical practices and counterfeit drugs. U.S.-European Union Mutual Recognition AgreementThis agreement would provide for reciprocal reliance on inspection systems in the United States and each of the 15 member nations of the European Union. The globalization of the pharmaceutical industry is outpacing our resources to inspect pharmaceutical manufacturing plants worldwide. Once fully implemented, the agreement would allow us to base our regulatory decisions on inspection data from "equivalent authorities" in the European Union. Equivalent authorities are those with regulatory systems for good manufacturing practices that we have assessed and determined will achieve a comparable level of public health protection. While the agreement would allow us to use an inspection report from one of our European counterparts as though it were our own, the actual regulatory decision would be up to us. Our experts in good manufacturing practices are leading the FDA team that is working with a team from the European Union to implement this agreement. A planned three-year transition to full implementation has ended, and two important issues remain to be resolved:
MRA update
IInternet sources
FDA/Center for Drug Evaluation and Research |