|
About CDER - Report |
U.S. Food and Drug Administration • Center for Drug Evaluation and Research |
|
About CDER - Report |
|
|
CDER Report to the Nation: 2002 Adobe Acrobat version of this document Drug Safety and QualityIndex
MissionProtect the public health by ensuring that human drugs are safe and effective. HighlightsThe practical size of premarketing clinical trials means that we cannot learn everything about the safety of a drug before we approve it. Therefore, a degree of uncertainty always exists about the risks of drugs. This uncertainty requires our continued vigilance, along with that of the industry, to collect and assess data during the post-marketing life of a drug. We monitor the quality of marketed drugs and their promotional materials through product testing and surveillance. As Americans are increasingly receiving the benefits of important new drugs before they are available to citizens of other countries, we must be especially vigilant in our surveillance. In addition, we develop policies, guidance and standards for drug labeling, current good manufacturing practices, clinical and good laboratory practices and industry practices to demonstrate the safety and effectiveness of drugs. Highlights of drug safety and quality activities in 2002 include:
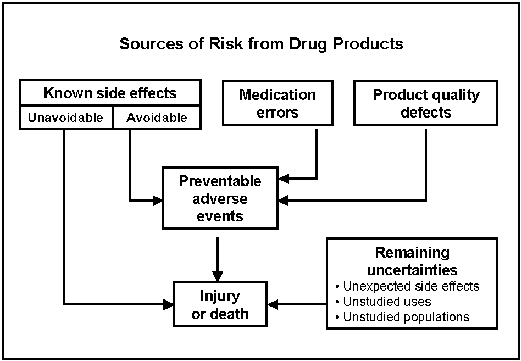
Types of Risks from Medicines
Product quality defects. These are controlled through good manufacturing practices, monitoring and surveillance. Known side effects. Predictable adverse events are identified in the drug's labeling. These cause the majority of injuries and deaths from using medicines. Some are avoidable, and others are unavoidable.
Medication errors. For example, the drug is administered incorrectly or the wrong drug or dose is administered. Remaining uncertainties. These include unexpected side effects, long-term effects and unstudied uses and populations. For example, a rare event occurring in fewer than 1 in 10,000 persons won't be identified in normal premarket testing. Medication error preventionWe work hard to ensure the safe use of drugs we approve by weeding out brand names that look or sound like the names of existing products. We identify and avoid brand names, labels and packaging that might contribute to problems or confusion in prescribing, dispensing or administering. We review about 250 reports of medication errors each month. About half are due to error-prone labeling such as look-alike labels, poor package design and confusing names. Our comprehensive Web site on medication errors is at http://www.fda.gov/cder/drug/MedErrors/default.htm. Drug SafetyWe evaluate the ongoing safety profiles of drugs available to American consumers using a variety of tools and disciplines. We maintain a system of postmarketing surveillance and risk assessment programs to identify adverse events that did not appear during the drug development process. We monitor adverse events such as adverse reactions, drug-drug interactions and medication errors. We have access to commercial databases that contain non-patient-identifiable information on the actual use of marketed prescription drugs in adults and children. This dramatically augments our ability to determine the public health significance of adverse event reports we receive. As we discover new knowledge about a drug's safety profile, we make risk assessments and decisions about the most appropriate way to manage any new risk or new perspective on a previously known risk. Risk management methods may include new labeling, drug names, packaging, "Dear Health Care Practitioner" letters, education or special risk communications, restricted distribution programs or product marketing termination. Adverse Event Reporting SystemA powerful drug safety tool is the Adverse Event Reporting System. This computerized system combines the voluntary adverse drug reaction reports from MedWatch and the required reports from manufacturers. These reports often form the basis of "signals" that there may be a potential for serious, unrecognized, drug-associated events. When a signal is detected, further testing of the hypothesis is undertaken using various epidemiological and analytic databases, studies and other instruments and resources. AERS offers paper and electronic submission options, international compatibility and pharmacovigilance screening.
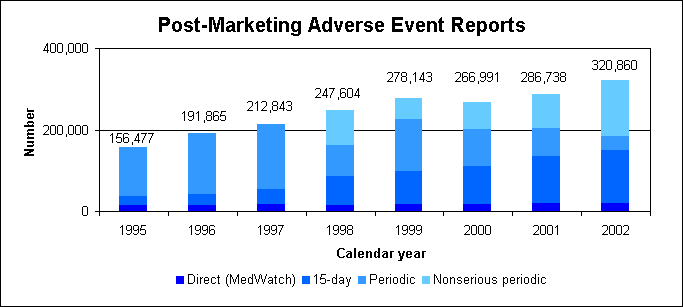
Adverse event reportingIn 2002, we received 320,860 reports of suspected drug-related adverse events:
Report types
Electronic submissionsAERS was designed and implemented so that the majority of the reports would be entered electronically. We are in the process of migrating the reporting format from paper to electronic. In a pilot program, we are accepting electronic individual case safety reports from five major drug firms. Electronic submissions into AERS represent 15 percent of the total expedited reports we received. We estimate the cost of receiving a report is cut from $31 per report to $3 to $19 per report for those submitted electronically. AERS on InternetYou can learn more about the Adverse Event Reporting System at http://www.fda.gov/cder/aers/default.htm. Adverse event reporting enforcementWe enforce regulations on postmarketing adverse event reporting to ensure that reports are accurate, timely and complete. We develop regulatory strategies and initiate inspections to determine industry compliance with the regulations. We use a risk-based approach to identify firms for inspection. We focus on firms with:
MedWatch Outreach and ReportingWe administer the MedWatch program that helps promote the safe use of drugs by:
Last year, subscribers to our e-mail notification service increased to about 30,000. We issued 36 safety alerts for drugs. Notifications were posted on the Internet and e-mailed to individuals and our 190 MedWatch partner organizations. Each month, our subscribers and partners received 25 to 45 safety-related labeling changes for drugs. MedWatch drug safety Internet resourcesThe latest medical product safety information can be found at http://www.fda.gov/medwatch/. You can sign up for immediate e-mail notification of MedWatch safety information at http://www.fda.gov/medwatch/new.htm. We may require specific written patient information for selected prescription drugs that pose a serious and significant public health concern. This information is called a Medication Guide. Medication Guides must be distributed to patients with each prescription dispensed. We require Medication Guides when the information is necessary for patients to use the product safely and effectively or to decide whether to use or to continue to use the product. Last year, we approved Medication Guides for four innovator products and one generic product:
Patient information for prescription drugs We continued our research and evaluation activities in support of the private sector providing patients with useful information about their prescription drugs. The target goal for 2006 is for 95 percent of patients to receive useful information with new prescriptions. This past year we worked on evaluating the written patient medication information materials they received. Additionally, we carried out a telephone survey of U.S. consumers about where they get their information about prescription drugs. Drugs with special safety restrictionsControls on 10 prescription drugs include limiting distribution to specific facilities; limiting prescription to physicians with special training or expertise; or requiring certain medical tests with their use. Consumers should not buy these drugs over the Internet. As of April 30, 2003, these drugs are:
More information is at http://www.fda.gov/oc/buyonline/consumeralert120902.html. Drug ShortagesWe work to help prevent or alleviate shortages of medically necessary drug products. Drug shortages occur for a variety of reasons including manufacturing difficulties, bulk supplier problems and corporate decisions to discontinue drugs. Because drug shortages can have significant public health consequences, we work with all parties involved to make sure all medically necessary products are available within the United States. Drug shortage program aids counterterrorism effortUtilizing data obtained from manufacturers and distributors, our drug shortage program provides supply and production information in response to federal government requests in relation to counterterrorism efforts. Drug shortages on the InternetWe have a Web site that lists current drug shortages, describes efforts to resolve them and explains how to report them.
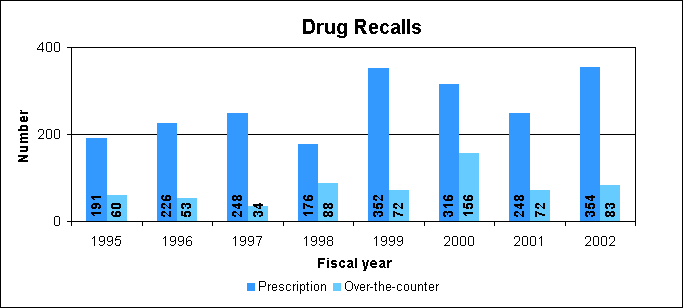
Drug Recalls and WithdrawalsIn some cases, a drug product must be recalled due to a problem occurring in the manufacture or distribution of the product that may present a significant risk to public health. These problems usually, but not always, occur in one or a small number of batches of the drug. The most common reasons for drug recalls include those listed in the column at the right. In other cases, a drug is determined to be unsafe for continued marketing and must be withdrawn completely. RecallsManufacturers or distributors usually implement voluntary recalls in order to carry out their responsibilities to protect the public health when they need to remove a marketed drug product that presents a risk of injury to consumers or to correct a defective drug product. A voluntary recall of a drug product is more efficient and effective in assuring timely consumer protection than an FDA-initiated court action or seizure of the product.
Drug recalls in fiscal year 2002
How we coordinate drug recallsWe coordinate drug recall information, assist manufacturers or distributors in developing recall plans and prepare health hazard evaluations to determine the risk posed to the public by products being recalled. We classify recall actions in accordance to the level of risk. We participate in determining recall strategies based upon the health hazard posed by the product and other factors including the extent of distribution of the product to be recalled. We determine the need for public warnings and assist the recalling firm with public notification about the recall. Top 10 reasons for drug recalls in fiscal year 2002:
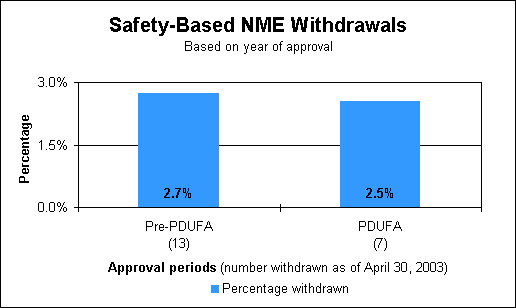
No safety-based withdrawals in 2002In some cases, there is an intrinsic property of a drug that makes it necessary to withdraw the drug from the market for safety reasons. There were no drugs withdrawn from the U.S. market last year for safety reasons. Record of safety-based market withdrawalsWhen drug withdrawals are compared based on year of approval, the recent period when we applied user-fee review goals is similar to the previous period.
Recent safety-based drug withdrawalsDrug name (year approved/ year withdrawn)
Risk management plan for alosetronImmediately after the announcement of the safety-related withdrawal of alosetron in 2000, distraught patients, stunned that this therapy had been taken away from them, began to contact us demanding access to a drug they characterized as "giving them their lives back." Alosetron, a treatment for diarrhea-predominant irritable bowel syndrome in women, had been withdrawn because outcomes from ischemic colitis, a known side effect, were more serious than predicted by the results of clinical trials and because of serious complications of constipation. We worked with the manufacturer to compile a formal risk management program for alosetron, which we approved last year. This program includes:
The indicated patient population was narrowed, and a lower starting dose was specified in the label. We have approved a number of programs designed to limit the risks of specific drugs. In the case of certain drugs such as thalidomide and clozapine, these programs are of proven benefit. Drug Promotion ReviewThe information about a drug available to physicians and consumers is just as important to its safe use as drug quality. We promote and protect the health of Americans by ensuring that drug advertisements and other promotional materials are truthful and balanced. We operate a comprehensive program of education, surveillance and enforcement about drug advertising and promotion.
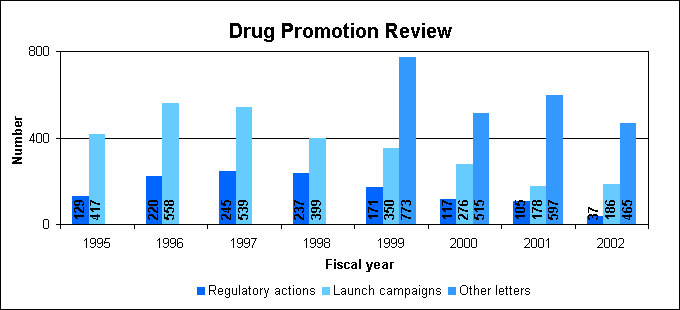
Drug promotion review statisticsWe issued a total of 688 drug promotion letters last year.
Launches and advisoriesWhen requested, we review advertisements and other promotional materials before drug companies launch marketing campaigns that introduce new drugs or campaigns that introduce new indications or dosages for approved drugs. In fiscal year 2002, we issued 186 advisory letters to companies regarding their promotional materials for launch campaigns. We issued 304 other advisory letters to the industry regarding proposed promotional pieces, both professional and consumer directed. In addition, we issued 161 other types of correspondence to the pharmaceutical industry, such as letters of inquiry, closure letters or acknowledgement letters. Regulatory actionsWe issued 37 regulatory action letters to companies for prescription drug promotions determined to be false, misleading, lacking in fair balance of risks and benefits or that promoted a product or indication before approval. These were either "untitled" letters for violations or "warning" letters for more serious or repeat violations. Examples of specific types of violative promotions include promotional exhibit hall displays, oral representations, Internet sites, plus traditional materials such as journal advertisements and sales brochures. We are also making sure that our warning and untitled letters will stand up in court, to provide more effective deterrence to recurrent patterns of misleading advertising. Direct-to-consumer promotionIncluded in our letters were 188 regarding direct-to-consumer promotion. This compares with 190 letters in 2001. Of last year's letters, 36 were for launch campaigns, 142 for non-launch advisories, and 10 were regulatory letters. We are working on improving our oversight of DTC advertising. Evidence from our studies as well as those conducted by consumer groups and other entities consistently shows that DTC ads lead to more patients seeking care for undertreated conditions. This often results in a different treatment that is more appropriate for the patient than the advertised drug. But physicians and others are concerned that consumers may not always get a balanced view of the benefits and risks of a product. Consequently, we are working on the issue of how manufacturers could provide clearer and more concise information for consumers derived from the drug's approved labeling. DTC advertising surveysWe completed two national telephone surveys and conducted preliminary analyses. One survey of 943 consumers is a follow-up to the 1999 survey of patients' attitudes and behaviors associated with direct-to-consumer advertisements. The other is a new survey of 500 physicians' attitudes and behaviors associated with direct-to-consumer advertisements. Preliminary findings of the two surveys include:
More is available at http://www.fda.gov/cder/ddmac/globalsummit2003/index.htm. Proposed rule to revise prescription drug labelingWe continued to work on a final rule, based on comments from the public to our proposal in 2001. The main purpose of labeling is to communicate essential information about prescription drugs to health care providers. Drug Product QualityWe provide comprehensive regulatory coverage of the production and distribution of drug products. We manage inspection programs designed to minimize consumer exposure to defective drug products. We have two basic strategies to meet this goal:
We identify, evaluate and analyze inspection findings for trends in deficiencies. We develop guidances to assist drug manufacturers in gaining a better understanding of our regulations. We communicate the expectations of compliance through outreach programs. We review all international pharmaceutical inspection reports. We determine which foreign manufacturers are acceptable to supply active pharmaceutical ingredients or finished drug products to the U.S. market. Reporting systems for drug quality problemsTwo important post-marketing tools help us rapidly identify significant health hazards associated with the manufacturing and packaging of drugs:
Manufacturing plant inspectionsFDA field offices conduct inspections of domestic and foreign plants that manufacture, test, package and label drugs. Before a drug is approved, FDA investigators must determine if data submitted in the firm's application are authentic and if the plant is in compliance with good manufacturing practices. After a drug is approved, FDA conducts an inspection to make sure a firm can consistently manufacture the product. Finally, routine inspections evaluate the firm's entire operations.
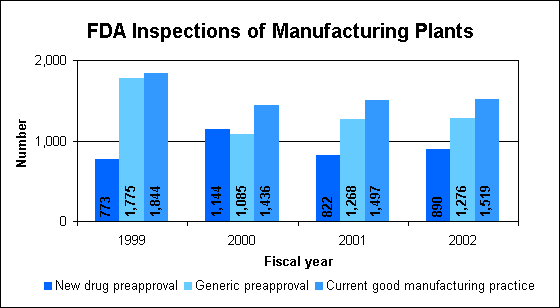
Preapproval inspectionsDuring fiscal year 2002, FDA evaluated:
Good manufacturing practice inspectionsThere were 1,519 good manufacturing practice inspections (1,109 non-gas) in fiscal year 2002.
Misbranded drugs, unsubstantiated claims Mislabeled, fraudulent, hazardous products. We often encounter mislabeled and fraudulent products that make unsubstantiated claims. Consumers may use these products inappropriately or incorrectly. They may use a fraudulent product for treating a serious disease condition in place of an effective treatment or delay the use of effective treatment. For these reasons, products that are mislabeled, fraudulent or make unproven claims may pose a significant health risk. Occasionally, fraudulent products may also contain toxic compounds that are likely to cause serious illness or injury. In addition, the marketing of products that are either mislabeled or fraudulent threatens to undermine the U.S. drug development and approval process as well as the ongoing over-the-counter drug review process. Protecting consumers from misbranded or fraudulent drugsWe protect consumers from mislabeled, fraudulent or hazardous products by identifying and taking steps to remove products that pose public health risk from the market. We issue enforcement letters and pursue enforcement actions such as seizures of violative products and injunctions against firms or individuals. Last year, more than 30 such letters were issued based on misbrandings or fraudulent claims found in labeling and on Internet sites. These addressed various illegally marketed products, many masquerading as dietary supplements, including several containing androstendione, ephedra-related compounds or human growth hormone. We also sent letters concerning "street drug alternatives," products intended for recreational use as alternatives to illicit controlled substances. Prescription drugs sold without approved applicationsWe identify drugs that are marketed without an approved new or generic drug application. We assess unapproved drugs to maximize protection of the public health and make best use of FDA's limited resources. We prioritize drugs that may be subject to compliance actions into risk-based categories according to safety considerations, effects on public health or subversion of the new drug approval process. Risk-based surveillance sampling of drugsWe monitor the quality of the nation's drug supply through surveillance and sampling of foreign and domestic finished dosage forms and bulk shipments of active ingredients. The drug products surveyed are selected according to a risk-based strategy that targets products with the greatest potential to harm the public health. FDA district offices conduct follow-up inspections to determine the cause of sample failures and to assure corrective action by the firms. Sampling criteria
Drug Product Quality ScienceLaboratory support Last year our efforts included:
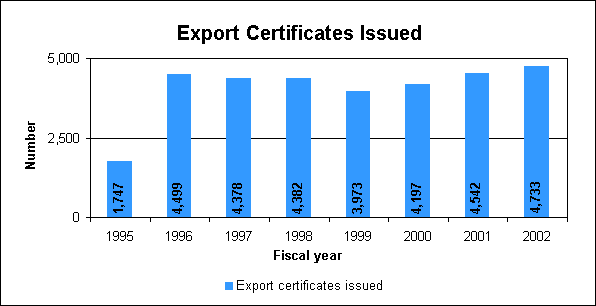
Process analytical technologies initiative Our goal for this initiative is to facilitate the introduction of new and emerging technologies that will improve the capability and efficiency of the pharmaceutical manufacturing process while maintaining or improving product quality. Known as process analytical technologies, these are systems for continuous analysis and control of manufacturing processes based on real-time or rapid measurements during processing. These systems involve in-line, on-line or at-line monitoring, measuring and controlling in manufacture of drug substance and drug products. We are using a collaborative process to develop this initiative. We are bringing together experts in the areas of analytical chemistry, physical chemistry, pharmaceutical technology, regulatory compliance, chemical engineering and international pharmaceutical manufacturing. These include experts from industry and academia along with our own and those from other FDA components. We are encouraging the adoption of this technology in drug manufacturing because it can enhance process understanding, improve overall product quality and lead to increased efficiencies. This also addresses many of the objectives of the Pharmaceutical cGMPs for the 21st Century Initiative. A steering committee comprised of senior FDA managers is involved in the development of a general guidance on the use of these new technologies. We have formed a special review team to evaluate process analytical technologies when used by the industry. On the team, our own chemistry reviewers and compliance officers will join FDA's field investigators on inspections. By organizing public meetings and workshops, we have gathered information related to development and use of process analytical technologies and shared our own research data. MicrobiologyWe assess product sterility, maintenance of product safety and the microbiological controls used by firms for drug development and manufacturing. Our microbiology review assures the safety of sterile and non-sterile products through scientific evaluation and communication with the industry and assures consistency through guidance documents. We promote the development of uniform and practical test methods and criteria for our own use and through the U.S. Pharmacopoeia and the International Conference on Harmonization (page 38). We heave a new program to advance rapid microbiology test methods. Export CertificatesWe promote goodwill and cooperation between the United States and foreign governments through the Export Certificate Program. These certificates enable American manufacturers to export their products to foreign customers and foreign governments. The demand for certificates by foreign governments remains high due to expanding world trade, ongoing international harmonization initiatives and international development agreements.
Export certificates issued in fiscal year 2002: 4,733The certificates attest that the drug products are subject to inspection by the FDA and are manufactured in compliance with current good manufacturing practices. Export certificates verify that drug products being exported:
FDA/Center for Drug Evaluation and Research |