|
Guidance for Industry
Nonclinical Safety Evaluation of Drug or Biologic Combinations
U.S. Department of Health and Human Services
Food and Drug Administration
Center for Drug Evaluation and Research (CDER)
March 2006
Pharmacology and Toxicology
Additional copies are available
from:
Office of Training and
Communications
Division of Drug Information, HFD-240
Center for Drug Evaluation and Research
Food and Drug Administration
5600 Fishers Lane
Rockville, MD 20857
(Tel) 301-827-4573
http://www.fda.gov/cder/guidance/index.htm
U.S. Department of Health and Human Services
Food and Drug Administration
Center for Drug Evaluation and Research (CDER)
March 2006
Pharmacology and Toxicology
TABLE OF CONTENTS
I.
INTRODUCTION
II.
NONCLINICAL STUDIES FOR A COMBINATION OF
two or MORE PREVIOUSLY MARKETED DRUGs OR BIOLOGICS (figure A)
A. Safety Considerations
B. Nonclinical Study
Recommendations/General Procedure
III.
NONCLINICAL STUDIES FOR A COMBINATION OF
DRUGs OR BIOLOGICS WHEN ONE OR MORE IS PREVIOUSLY MARKETED AND one
IS A new molecular entity (figure B)
A. General Toxicology
Studies
B. Reproductive and
Developmental Toxicology
C. Animal Models of
Efficacy
D. Further Studies
IV.
NONCLINICAL STUDIES FOR A COMBINATION OF TWO
OR MORE DRUGs or Biologics WHEN ALL ARE New molecular entities
(Figure C)
A. General Toxicology
Studies
B. Animal Models of
Efficacy
C. Safety Pharmacology
D. PK/ADME and
Toxicokinetics
E. Genetic Toxicology
F. Special Toxicology
G.
Reproductive and Developmental Toxicology
H. Further Studies
I. Carcinogenicity
Appendix: Recommended General Procedures
Figure A:
Combinations of Previously Marketed Drugs or Biologics
Figure B:
Combinations of Previously Marketed Drugs or Biologics with NMEs
Figure C:
Combinations of NMEs with NMEs
Guidance for Industry
Nonclinical Safety Evaluation of
Drug or Biologic Combinations
This
guidance represents the Food and Drug Administration’s (FDA’s)
current thinking on this topic. It does not create or confer any
rights for or on any person and does not operate to bind FDA or
the public. You can use an alternative approach if the approach
satisfies the requirements of the applicable statutes and
regulations. If you want to discuss an alternative approach,
contact the FDA staff responsible for implementing this guidance.
If you cannot identify the appropriate FDA staff, call the
appropriate number listed on the title page of this guidance.
This
guidance provides recommendations on nonclinical approaches to
support the clinical study and approval of fixed-dose
combination products (FDCs), co-packaged products, and some
adjunctive therapies.
The intent of this guidance is
to delineate general guiding principles. To receive more detailed
advice regarding a particular drug or biologic combination
development program, sponsors should contact the appropriate review
division before submitting an investigational new drug application (IND).
In addition, the Food and Drug Administration (FDA) is in the
process of publishing more specific guidance for certain categories
of drug combinations.
Drug and biologic combinations may involve: (1)
two or more previously marketed drugs or biologics
(MD/Bs); (2) one or more new molecular entities (NMEs) and one or
more previously marketed drugs or biologics; or (3) more than one
NME. The nonclinical studies considered important for each type of
combination may differ, depending upon the information available on
each drug substance. The nonclinical studies that the FDA
recommends sponsors use to characterize the combination will depend
on the toxicologic and pharmacokinetic profiles of the individual
drugs or biologics, the treatment indication or indications, and the
intended population. The number and type of studies will depend on
the stage of clinical development.
In this guidance, each of the three general
types of combinations (i.e., MD/B-MD/B, MD/B-NME, and NME-NME) will
be discussed separately. This guidance covers combinations of drugs
and biologics regulated by the Center for Drug Evaluation and
Research (CDER). The ICH guidance for industry S6 Preclinical
Safety Evaluation of Biotechnology-Derived Pharmaceuticals
should also be consulted for nonclinical development of biologic
products.
FDA’s guidance documents, including this
guidance, do not establish legally enforceable responsibilities.
Instead, guidances describe the Agency’s current thinking on a topic
and should be viewed only as recommendations, unless specific
regulatory or statutory requirements are cited. The use of the word
should in Agency guidances means that something is suggested
or recommended, but not required.
This section addresses the situation in which a
sponsor submits an application to develop a combination of two or
more previously marketed drugs or biologics or a combination of
drugs and biologics. Generally the FDA believes that, in such a
situation, sufficient clinical and nonclinical data will exist for
each drug product separately. However, the indications for which
each drug is marketed should be compared to those for which the
combination is being proposed. For example, drug products marketed
for acute use may not have nonclinical data to support chronic use.
To the extent that there are gaps in the data, the FDA may recommend
that additional nonclinical studies be conducted.
If existing clinical and nonclinical safety
data for each separate drug or biologic are sufficient to support
the safety of the proposed new indication, then the FDA recommends
that the following factors relevant to the safety of the combination
be considered to determine whether further nonclinical studies are
warranted.
- Information available on prior human
experience with the combination. The FDA recommends that the
sponsor provide a summary of the available data in humans (if any)
on the use of the combination. The FDA also encourages the
sponsor to provide copies of any relevant published studies in
humans (or animals). Such reports may not provide definitive
safety data, but they may provide some measure either of assurance
or reasons for concern.
- Information known about the individual drugs
or biologics in animals and humans and concordance of
pharmacokinetics (PK), pharmacodynamics (PD), and toxicologic
effects in animals with the analogous data for humans.
- Possibility of a PD interaction.
Drugs/biologics may exhibit affinity for the same receptors or
biologic targets or may produce similar effects on physiologic
function, related or not to their mechanism of action.
- Possibility of a PK interaction. A PK
interaction can manifest in several ways, some of which can be
monitored in vivo and some of which cannot. One drug/biologic
product may alter the absorption or excretion of another product,
change its distribution into one or more tissues, or change its
pattern or rate of metabolism. Drugs may compete for serum
protein binding, resulting in an increase in circulating free
levels and tissue uptake of one drug.
- Possibility of a toxicologic interaction
(i.e., that the target organs for toxicity are similar for each
drug/biologic). This situation may result in a lowering of the
previously determined no-effect doses for one or both
drug/biologic products and more severe toxicities in the affected
organs. The FDA will consider all known toxicology on the product
(e.g., general toxicity, reproductive toxicity, carcinogenicity,
and safety pharmacology studies (cardiovascular, central nervous
system (CNS), respiratory)).
- Margin of safety for each drug/biologic
product. If one or more of the drugs/biologics has a narrow
margin of safety (i.e., causes serious toxicity at exposures close
to the predicted clinical exposure), then the possibility of drug
interaction is of particular concern, especially if the toxicity
is not reversible or cannot be monitored clinically.
- Possibility that the drugs/biologics compete
for or alter the activity or endogenous levels of the same enzymes
or other intracellular molecules (e.g., co-administration of two
pro-oxidants could deplete endogenous levels of glutathione).
- Possibility of a chemical interaction. One
drug may chemically modify another drug or biologic (e.g., one
drug may oxidize, methylate, or ethylate the other drug or
biologic). This could result in NMEs with new toxicities.
- Possibility that one drug/biologic may
compromise the effectiveness of another drug/biologic for a
lifesaving therapy.
If existing clinical and nonclinical safety
data for each separate drug or biologic are sufficient to support
the safety of the proposed new indication, including the dose,
dosing schedule, duration, and new patient population, then
additional nonclinical studies may not be needed.
The general approach to addressing the safety
concerns posed by the testing or marketing of combinations of
previously marketed drugs or biologics is illustrated in Figure A.
The safety of the combination should be assessed according to the
factors listed in section II.A (see Figure A, Boxes 1 to 2). If
neither individual drug or biologic product has serious toxicity at
exposures well above the proposed clinical exposure or if there is
substantial clinical experience with the combination, the FDA may
recommend that additional nonclinical studies do not need to be
conducted before testing in humans, during initial studies in
humans, or at all (Boxes 2 to 3). The Agency’s
recommendation to conduct nonclinical studies for further
development of the combination will depend on what is learned from
initial studies in humans or what is known from prior human use of
the combination.
If after evaluating the available data on the
individual drug/biologic products and the potential for
drug/biologic interaction there is no evidence to suggest a possible
interaction, direct assessment of the combination by testing in
animals may not be needed before the first small clinical studies
with the combination. Even if an interaction is expected,
nonclinical studies may not be necessary if the expected interaction
is likely to result in predictable, nonserious, monitorable effects
in humans. For example, if a metabolic interaction is predicted,
the starting dose could be significantly lowered in humans.
Metabolic interactions have been seen for combinations of two
biologics. There is a possibility of drugs/biologics affecting the
same tissue or biologic target or having a PD interaction.
Generally, the FDA recommends that sponsors
conduct nonclinical toxicity studies before clinical studies are
initiated if: (1) the drug products have similar target organ
toxicity or PD activity; and (2) either drug product causes serious
or nonmonitorable toxicity in animals or humans at exposures near
the clinical exposure; or (3) any other reason exists for serious
clinical concern (see section II.A). The particular nonclinical
studies recommended by the FDA will depend on a number of factors,
including the nature of the toxicity and on the concerns identified
in section II.A. For assessment of general toxicity, a bridging
study may be appropriate, provided the duration is sufficient to
elicit the toxicity of concern. For example, a general toxicity
bridging study of 3 months’ duration could be considered for a
chronic indication. The FDA suggests that combination studies
include an assessment of several dose levels of the combination and
a high dose of each drug alone. Other possible designs can be
discussed with the review division. Sponsors are urged to select
the doses of each drug used in combination to allow for additive or
synergistic effects without unacceptable toxicity in the high-dose
groups. Usually, assessment of the drug combination may be
conducted in only one species if one of the following conditions
exists: (1) toxicity in a particular species has high concordance
with human toxicity or the toxicities are similar among species; or
(2) one species is a more relevant model for human risk based on
other factors such as PK/ADME (absorption, distribution, metabolism,
and excretion) or expression of the pharmacologic activity. If a
sponsor will be conducting only one general toxicity study, the FDA
recommends that the sponsor provide justification for the species
selected for testing the combination. There may be cases, however,
in which the Agency may recommend conducting studies in two species
despite one or both of these conditions being met. For example,
depending on the results in the first species, a new cause for
concern might warrant follow-up studies in a second species, if
there is an appropriate second species.
Sometimes one of the drugs proposed for the
combination will be much more toxic in animals than in humans, such
that animals cannot tolerate the combination at doses that produce
exposure relevant to the anticipated clinical exposure (e.g., some
nonsteroidal anti-inflammatory drugs (NSAIDs) and antibiotics). In
those cases, general toxicity studies of the combination could be
conducted at a dose giving less exposure than that achieved with the
recommended clinical dose of the more toxic drug product, provided
that a serious dose-limiting toxicity is achieved in the animals.
The timing of the studies should be as described in the ICH guidance
for industry M3 Nonclinical Safety Studies for the Conduct of
Human Clinical Trials for Pharmaceuticals.
Combination genotoxicity studies generally will
not be necessary if the individual agents have been tested
consistent with current standards. Embryo-fetal development studies
of the combination should be conducted per the timing
described in ICH M3, unless the marketed products are already known
to have significant risk for developmental toxicity (e.g., one of
the marketed drugs has been assigned a pregnancy category “D” or
“X”). If combination studies are needed, a single study could be
conducted in the most appropriate species, based on what is known
about the individual drugs or biologics. If a significant risk is
only identified for a particular trimester of pregnancy, such as ACE
inhibitors during the third trimester, studies to evaluate the
effects of exposure to the NME or the combination during other
trimesters may be needed.
For chronic indications, a carcinogenicity
study on the drug combination generally will only be recommended if
statistically significant incidences of preneoplastic lesions were
observed at a new organ or tissue site in nonclinical studies of the
combination. Results of the nonclinical studies may be used to
recommend modification of the clinical protocol (e.g., starting
clinical doses, parameters to monitor) (Box 8).
This section addresses the situation in which a
sponsor submits an application to develop a combination of two or
more drugs or biologics — one or more previously marketed and one an
NME or a combination of drugs and biologics.
The Agency generally suggests that nonclinical
studies be conducted on the NME for a product that is a combination
of an NME and a previously marketed drug or biologic. The FDA
believes that the standard battery of nonclinical studies (i.e.,
genetic toxicology, pharmacology, safety pharmacology, PK/ADME,
general toxicity, reproductive and developmental toxicity,
carcinogenicity) generally will be appropriate for the NME, as
described in ICH M3. ICH S6 should be consulted for nonclinical
development of biologic products. If genotoxicity studies on the
previously marketed product are consistent with current standards,
it may be appropriate to conduct genotoxicity studies on only the
drug NME portion of the combination.
Depending on the duration of the proposed
therapy, the FDA recommends that a sponsor conduct a bridging study
of up to 90 days with the combination in the most appropriate
species. Studies of shorter duration could be appropriate for
shorter clinical studies or for nonchronic indications, per ICH M3.
There may be cases, however, where studies in a second species may
be appropriate. Because the drug ratio may change during drug
development, it is important to design the toxicity studies to
provide adequate margins of safety for future clinical studies. For
combinations, the FDA recommends that the exposure to the
drugs/biologics be at ratios that are relevant to the intended
clinical use.
Sometimes one of the drugs proposed for the
combination will be much more toxic in animals than in humans, such
that animals cannot tolerate the combination at doses that produce
exposure relevant to the anticipated clinical exposure (e.g., some
NSAIDs and antibiotics). In those cases, nonclinical studies of the
combination could be conducted at a dose giving less exposure than
that achieved with the recommended clinical dose of the more toxic
drug product, provided that a serious dose-limiting toxicity is
achieved in the animals.
Embryo-fetal development studies on the NME are
recommended, as are fertility and other reproduction studies, by the
ICH. Studies to evaluate the reproductive toxicity of the
combination are not generally needed. Embryo-fetal development
studies of the combination should be conducted unless the
marketed drug substance or the NME is already known to have
significant risk for developmental toxicity (e.g., the marketed drug
has been assigned a pregnancy category “D” or “X”). If combination
studies are needed, a single study could be conducted in the most
appropriate species, based on what is known about the individual
drugs or biologics. If a significant risk is only identified for a
particular trimester of pregnancy, such as ACE inhibitors during the
third trimester, studies to evaluate the effects of exposure to the
NME or the combination during other trimesters may be needed.
Animal models of efficacy
are not generally needed. However, valuable data may be
obtained from studying the combination in appropriate animal models
of efficacy, if they are available and considered relevant. For
example, there are situations in which one drug has been shown to
alter the efficacy of the second drug. This information may be
important if one or more of the drugs in the combination is for a
serious or life-threatening indication. Identifying such an
interaction permits an informed choice of dose of each product in
the combination and an optimal schedule for the proposed clinical
trial.
The FDA recommends that a sponsor address any
important data gaps for the marketed product or products that may be
relevant for the proposed indication. After evaluating the
available data on the individual drug products and the data on the
bridging study of up to 90 days on the combination, a determination
will be made on whether it is appropriate to conduct additional
studies to address potential drug/biologic interactions. If a drug
interaction is identified in the bridging study (synergistic
effects) and the mechanism (e.g., PK, PD, or overlapping toxicity)
is not apparent, then the FDA urges sponsors to consider studies to
understand the nature of the interaction. The possible mechanisms
of drug interaction listed in section II.A would also apply to
combinations of one or more previously marketed drugs/biologics and
an NME. Other than the general toxicology bridging study of up to
90 days and studies on embryo-fetal development, additional studies
on the combination generally will not be needed.
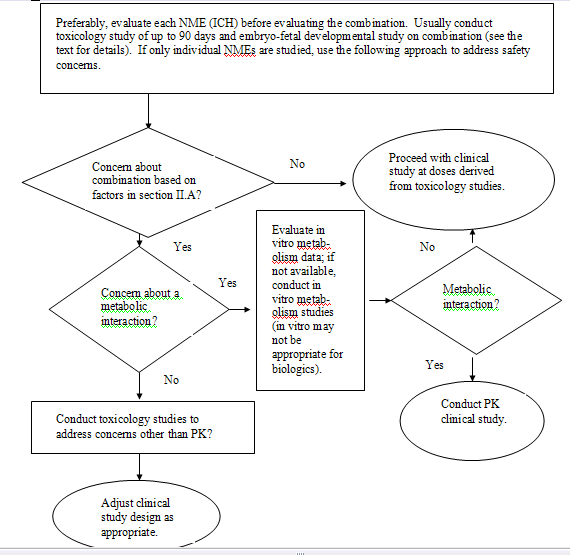
The FDA generally recommends that the sponsor
conduct nonclinical studies on each NME to evaluate the safety of a
combination of NMEs. Sponsors are encouraged to conduct the
standard battery of nonclinical studies (i.e., genetic toxicology,
pharmacology, safety pharmacology, PK/ADME, general toxicity,
reproductive and developmental toxicity, carcinogenicity) on each
NME, according to the timing as described in ICH M3, and according
to ICH S6 for nonclinical development of biologic products.
Depending on the duration of the proposed therapy, a bridging study
of up to 90 days (for chronic indications) should be conducted with
the combination in the most appropriate species if the NMEs were
evaluated as separate entities (which is preferred) and not as a
combination. There may be cases, however, where studies in a second
species may be appropriate. If the two drugs or biologics are
proposed to be marketed together only, then it is possible that it
may be sufficient to conduct toxicology studies only on the
combination. However, nonclinical studies conducted on each NME
alone can be invaluable should it become important to alter the
clinical regimen from what is initially proposed or studied.
Because the drug ratio may change during drug
development, it is important to design the toxicity studies to
provide adequate margins of safety for future clinical studies. The
FDA recommends that the drugs or biologics be tested at doses that
produce exposure ratios that are relevant (i.e., somewhat similar)
to the intended clinical use, when feasible.
Sometimes one of the drugs proposed for the
combination will be much more toxic in animals than in humans, such
that animals cannot tolerate the combination at doses that produce
exposure relevant to the anticipated clinical exposure (e.g., some
NSAIDs and antibiotics). In those cases, nonclinical studies of the
combination might be conducted at a dose giving less exposure than
that achieved with the recommended clinical dose of the more toxic
drug, provided that a serious dose-limiting toxicity is achieved in
the animals at the highest dose.
Animal models of efficacy are not generally
needed. However, valuable data may be obtained from studying the
combination in appropriate animal models of efficacy, if they are
available and considered relevant. For example, there are
situations in which one drug has been shown to alter the efficacy of
the second drug. This information is especially important if one or
more of the drugs or biologics in the combination is for a serious
or life-threatening indication. Identifying such an interaction
permits an informed choice of dose of each product in the
combination and an optimal schedule for the proposed clinical trial.
The FDA strongly recommends that sponsors
assess the effects of drugs/biologics on a variety of organ systems
before dosing in humans. Combination safety pharmacology studies of
a particular organ system (e.g., cardiac, respiratory, CNS) may be
valuable in many situations, such as when both drugs target the same
organ system, a toxicity is associated with a class of compounds
(e.g., QT prolongation), or the intended patient population is
compromised (e.g., renal impairment). Sometimes when the molecular
target is known, effects of the combination can be predicted.
The FDA recommends that sponsors conduct
combination PK/ADME studies to assess the potential for a PK
interaction between the drugs/biologics. These data are valuable
for supporting the safety profile and guiding the drug/biologic
development process. The FDA further recommends that PK/ADME
combination studies (e.g., in vitro drug metabolism studies) be
conducted early in drug development. The FDA encourages sponsors to
evaluate serum protein binding and to monitor plasma concentrations
of each drug in the toxicology studies. It may be possible to
collect PK data as part of the toxicology studies instead of in a
separate study.
Genetic toxicology studies are not needed for
biologic or drug/biologic combinations. Assessing the genotoxic
potential of a drug/drug combination is generally not necessary,
provided that adequate studies of the individual drug substances
have been conducted. For the in vitro assays, genotoxic potential
is routinely tested in the absence and presence of metabolic
activation. Therefore, testing drugs in combination in these assays
would not likely provide additional information to assays testing
each drug alone, particularly if any potential interaction is
expected to be from effects on hepatic metabolism.
The FDA may recommend that a sponsor conduct
special toxicology studies, such as local tolerance studies, with
the NME as well as with the combination in a particular therapeutic
area relevant to the proposed use. The Agency may also recommend
that targeted toxicity studies be conducted, depending upon the
nature of toxicities seen in animals and humans with the drug
products or drug class.
Embryo-fetal development studies on each NME
are generally recommended by the ICH, as are fertility and other
reproduction studies. Studies to evaluate the reproductive toxicity
of the combination are not generally needed, if this has been
evaluated for the individual NME. If developmental toxicity has
been assessed only on each NME separately, then the FDA recommends
that developmental toxicity studies be conducted on the combination
as well, in the most appropriate species, before treatment of women
of childbearing potential. Embryo-fetal developmental studies of
the combination may not be needed if one of the NMEs is known from
the nonclinical studies to have significant risk for developmental
toxicity, such as results indicating potential labeling as a “D” or
“X.” If a significant risk is only indicated for a particular
trimester of pregnancy, studies to evaluate the effects of exposure
to the combination during other trimesters may be needed.
After evaluating the available data on the
individual drug/biologic products and the data on the bridging study
of up to 90 days on the combination, a determination will be made as
to whether it is appropriate to conduct additional studies to
address potential drug/biologic interactions. If a drug/biologic
interaction is identified in a study and the mechanism (e.g., PK,
PD, or overlapping toxicity) is not apparent, then the FDA urges the
sponsor to consider studies to evaluate the nature of the
interaction. The possible mechanisms of drug/biologic interaction
listed in section II.A would also apply to combinations of more than
one NME. Generally, studies of the combination other than the
general toxicology bridging study of up to 90 days (depending on the
chronicity of the indication) and studies on embryo-fetal
development will not be needed.
Depending on the
duration and intended use of the combination, the Agency may suggest
that the sponsor conduct carcinogenicity studies on the combination,
if each individual NME has not been tested for carcinogenicity.
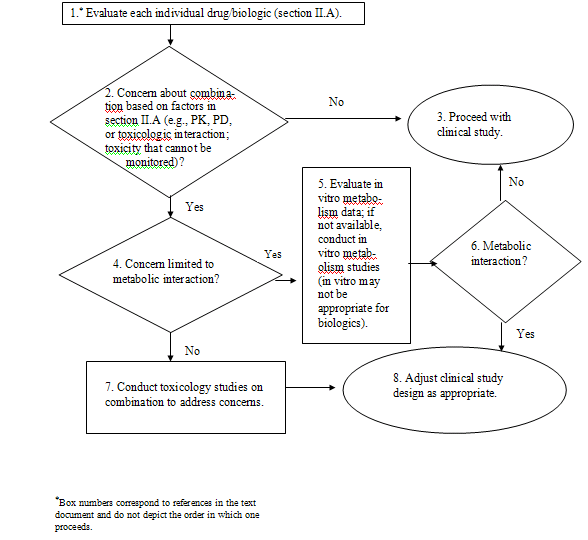
Figure B: Combinations of Previously Marketed Drugs or Biologics
with NMEs
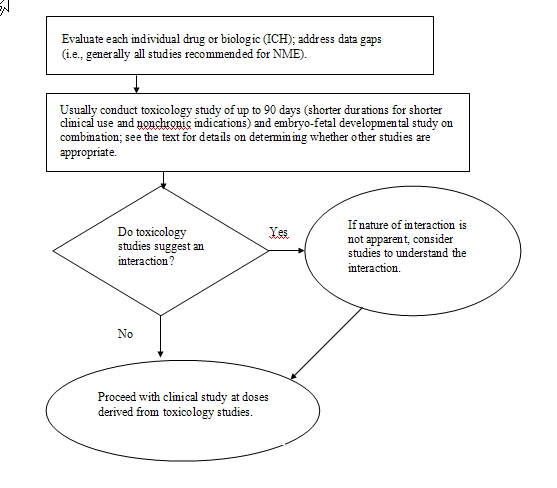
Figure C: Combinations of NMEs with NMEs
 Back
to Top Back
to Top  Back to Guidance Page
Back to Guidance Page
 PDF requires the free Adobe
Acrobat Reader PDF requires the free Adobe
Acrobat Reader
Date created: March 14, 2006 |



