 |
 |
 |
|
 |
FDA Home Page | Search
FDA Site | FDA A-Z Index | Contact
FDA | FDA Centennial
![]()
Gary Buehler, R.Ph
Director of the Office of Generic Drugs
Center for Drug Evaluation and Research
Food and Drug Administration
Special Committee on Aging
United States Senate
INTRODUCTION
Mr. Chairman and Members of the Committee, I am Gary Buehler, R.Ph, Director of the Office of Generic Drugs (OGD), in the Center for Drug Evaluation and Research (CDER), at the U.S. Food and Drug Administration (FDA or the Agency). Thank you for the opportunity to testify about FDA’s efforts to expedite the approval of generic drug products.
FDA understands that Congress and the public are concerned about the high cost of prescription drugs. Generic drugs play an important role in granting access to affordable products that will benefit the health of consumers, especially seniors – who often are on a fixed income. Prompt approval of generic drug product applications, also known as abbreviated new drug applications (ANDA), is imperative to making generic products available to American consumers at the earliest possible date.
Statutory Provisions
Prior to the passage of the Drug Price Competition and Patent Term Restoration Act (Hatch-Waxman Amendments) of 1984, FDA’s primary statute, the Federal Food, Drug, and Cosmetic (FD&C) Act, did not provide for the approval of generic drugs. The Hatch-Waxman Amendments established the ANDA approval process, which permits FDA to approve generic versions of previously approved innovator drugs without the submission of clinical studies and other kinds of data that are required in a full new drug application (NDA). An ANDA refers to the previously approved NDA of the innovator drug and relies upon the Agency’s finding of safety and effectiveness for that drug. Also, with respect to each unexpired patent submitted to FDA by the owner of the innovator drug and published by FDA in the Orange Book1 , an ANDA contains a certification that the ANDA applicant either will wait for the patent to expire before marketing the drug or that the applicant challenges the patent as invalid or not infringed.
The Hatch-Waxman Amendments have been very successful and have provided for the approval of over 8,000 generic drug products. These products are lower cost, high quality products that have saved the American public and the government billions of dollars.
FDA has taken a number of significant steps to provide greater access to affordable prescription medications, including unprecedented steps to lower drug costs by helping to speed the development and approval of low-cost generic drugs after legitimate patents have expired on branded drugs. Generic drugs typically cost 50 to 70 percent less than their brand-name counterparts. In 2003, FDA published a final rule to improve access to generic drugs and lower prescription drug costs for millions of Americans. This rule was first proposed in response, in part, to Federal Trade Commission recommendations and other changes the Agency identified as being useful in improving generic competition. The rule limits an innovator drug company to only one 30-month stay of a generic drug applicant’s entry into the market for resolution of a patent challenge. These changes will save Americans over $35 billion in drug costs over the next 10 years, and will also provide billions in savings for the Medicare and Medicaid programs. We were pleased that elements of this rule were codified as part of the Medicare law and that, with FDA’s technical assistance, the law added additional mechanisms to enhance generic competition in the marketplace.
In addition, since FY2001, the Administration and Congress have increased funding for FDA's generic drug program by 66 percent, a clear sign of the important role played by OGD. These increases have enabled FDA to hire additional expert staff to review generic drug applications more quickly and initiate targeted research to expand the range of generic drugs available to consumers. While there remains work to be done, as I will discuss, we have been able to produce significant reductions in approval times for generic drugs since 2002 that consequently will save consumers billions by generally reducing the time for developing generic drugs and making them available.
The Office of Generic Drugs’ Workload
Much concern has been raised from the public and Congress about a “backlog” of pending ANDAs, currently under OGD review. FDA has received an increased number of ANDAs in the last few years. OGD generally maintains a “first-in, first-reviewed” policy for ANDAs. FDA instituted this generic drug review priority to ensure the integrity of the approval process. A number of factors govern the timing of generic drug approvals, including: whether the application is of high quality, meets inspection standards and the scientific and technical requirements for approval, and whether patent protection and exclusivity periods have expired on the innovator drug.
There are several contributing causes to the increased number of generic applications FDA is receiving. Among these are the approvals of many new innovator drugs in the 1990s with patents that are now expiring, as well as the burgeoning number of new generic firms entering the market. Over the last five years, the number of applications submitted to OGD has increased by 150 percent. In fiscal year (FY) 2001, OGD received 307 ANDAs. In FY 2002 submissions increased 17.6 percent to 361. In FY 2003, they increased 24.3 percent to 449. In FY 2004, they increased 25.3 percent to 563. And, in FY 2005, they increased 36 percent to 766 applications submitted for review (see figure 1). Just last month, June 2006, we approved (or tentatively approved, meaning an application is technically ready for approval, but patent or exclusivity prevents immediate approval) 45 applications, however, the number of pending applications grew substantially because we received 92 applications. Clearly, this rate of increase in applications results in a dramatic increase in the workload for the review staff in OGD.
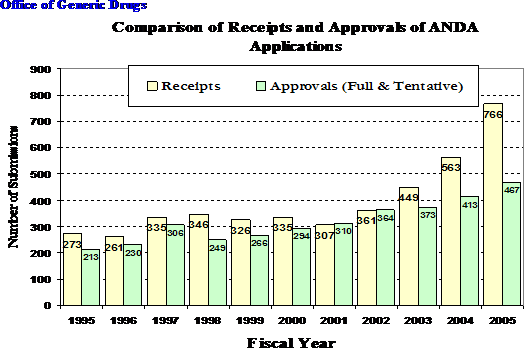
Figure 1
Although OGD still has a backlog, figure 1 also demonstrates that we have managed to increase the number of approvals each year. In FY 2001, OGD approved (or tentatively approved) 310 ANDAs and increased the annual number of approvals to 467 (or tentative approvals) in FY 2005. OGD’s efforts are also evident when looking at the median approval time. The median approval times have decreased from 18.4 months in FY 2001 to 16.3 months in FY 2005. In FY 2003, OGD approved (or tentatively approved) 132 applications in less than 15 months after receipt. In FY 2004, that number increased to 146 in less than 15 months and increased further to 174 in FY 2005 (102 of which were approved in less than 12 months). Despite these challenges, FDA has managed to maintain its rate of approval of more than one generic drug application a day.
It is important to understand that a pending ANDA has not been reviewed. When a pending ANDA is initially reviewed and deficiencies are communicated to the company, the application is no longer considered pending. However, when the company submits an amendment to its ANDA to address the identified deficiencies, the application is again considered pending. Therefore, the ANDAs in the backlog are not all unreviewed, but may be applications that have had an initial review and are now awaiting a second or subsequent review of the company’s attempts to satisfy approval requirements.
FDA has taken significant steps to improve our resources. Total spending on the Generic Drug Program is $64.6 million, which is more than a 66 percent increase from the comparable FY 2001 amount. FDA has increased its generic drugs full-time equivalent (FTE) positions from 134 in FY 2001 to 201 in FY 2006. Last year, FDA added 12 new FTE positions to OGD’s staff. These individuals, now fully trained, have recently reached the point in their learning curve where they are now full contributors to the efforts of OGD. In addition, OGD has taken actions to streamline the ANDA review process. These actions include adding a third chemistry review division and a fifth team in OGD’s Division of Bioequivalence. Also, a number of new review practices have been implemented to improve interactions with generic drug companies. We have begun utilizing non-reviewer Project Management staff to take certain actions not requiring scientific expertise, thus alleviating the burden of these activities on the review staff. OGD has instituted other efficiencies to application review. These include:
Because of these efforts, on the very day that the last patents or exclusivities expire on the innovator product, OGD has been able to approve at least one generic drug application in most cases. And, if there are no products eligible for 180-day exclusivity, we have usually been able to approve two or more applications for the same products. In fact, very recently, FDA approved generic applications for pravastatin (Pravachol), sertraline (Zoloft), and simvastatin (Zocor) when the innovator protections expired. Many Americans use one of these drugs. The availability of generic versions of these three drugs should produce savings measured in the billions of dollars per year. We will work to continue our success so far in staying ahead of the curve on first-time generics and responding to pending applications.
Citizen Petitions
FDA regulations permit any interested person to file a citizen petition requesting FDA “to issue, amend, or revoke a regulation or order, or to take or refrain from taking any other form of administrative action” (Title 21, Code of Federal Regulations 10.25 and 10.30). Citizen petitions may be submitted at any time, requesting that FDA impose new criteria for approval of ANDAs. The petitions often make serious challenges to whether or not a generic product can be approved; that is, whether a specific application or a group of applications would meet the statutory requirements for approval.
It is incumbent upon FDA to consider and address the merits of petitions. The data and information submitted with these petitions require detailed analysis and precise scientific documentation, often involving multiple disciplines within CDER. Because the same issues sometimes are raised in a subsequent court challenge to an ANDA approval and because petitioners sometimes submit non-scientific petitions that raise purely legal questions related to ANDA approvals, a thorough legal review is also necessary. Although it is not required that a citizen petition response be issued before approval of a related ANDA, it is important that FDA comprehensively assess the scientific issues prior to approval of the ANDA. It is very rare that petitions present new issues that CDER has not fully considered, but the Agency must nevertheless assure itself of that fact by reviewing the citizen petitions.
A high percentage of the petitions OGD reviews are denied. An analysis of petitions answered between calendar years 2001 and 2005, raising issues about the approvability of generic products (42 total responses), showed that FDA denied 33, denied three in part, and granted six. It should be noted that when petitions are granted, wholly or in part, it is often because FDA already has the proposed scientific or legal standard in place or is already planning to take the action that the petition requests. While the citizen petition process is a valuable mechanism for the Agency to receive information from the public, it is noteworthy that very few of these petitions on generic drug matters have presented data or analysis that significantly altered FDA’s policies. Of the 42 citizen petition responses examined, only three petitions led to a change in Agency policy on the basis of data or information submitted in the petition.
CDER has made considerable efforts in the last year-and-a-half to improve the process for responding to citizen petitions. As part of this process, OGD constituted a group of highly qualified and skilled scientists dedicated to assessing the citizen petitions related to generic drugs and formulating FDA’s responses to them. Other improvements include: increased prospective management of the petition response process; development of clear timelines for completing actions; and improved communication among the CDER components involved in responding to citizen petitions.
Authorized Generics
The term “authorized generic” is generally used to describe an instance when an innovator company, in the face of pending generic competition, repackages its own product and markets it as a “generic.” Prior FDA approval is not needed for the innovator company to do this, as review and approval occur under the auspices of the innovator’s approved NDA. Generic drug companies, through citizen petitions and lawsuits, have sought FDA’s intervention to halt the marketing of authorized generics. FDA determined, and the courts upheld, that the FD&C Act does not give FDA authority to intervene in the matter.
CONCLUSION
FDA appreciates the Committee’s interest and concern about expediting the approval of generic drug products and the opportunity to discuss these important issues. I am constantly impressed by the dedication, skills and effectiveness of FDA staff responsible for reviewing generic drugs. In spite of a tremendous workload, be assured that there is a sense of purpose and knowledge, among my staff and this Administration that they are working towards an important public health mission. FDA will continue to work towards greater efficiency in ANDA review and attempt to deal with the issues discussed today and the many emerging challenges ahead. We are committed to continue to make additional generic products available to the American public as soon as legally possible. I would be pleased to respond to questions.
1 The publication, “Approved Drug Products with Therapeutic Equivalence Evaluations” (commonly known as the Orange Book) identifies drug products approved on the basis of safety and effectiveness.
![]() Get free
weekly updates about FDA press releases, recalls, speeches, testimony
and more.
Get free
weekly updates about FDA press releases, recalls, speeches, testimony
and more.
![]()