 |
Review of Therapeutic Equivalence
Generic Bupropion XL 300 mg and Wellbutrin XL 300 mg
Between January 1 and June 30, 2007, FDA received 85 post-marketing reports in which patients who switched from Wellbutrin XL 300 mg to Teva’s bupropion formulation (Budeprion XL 300 mg) experienced an undesirable effect. Specifically, in 78 of these cases, there was a reported loss of antidepressant effect following a switch from the branded to generic product. In addition to the loss of effect, a number of cases also reported the new onset or worsening of side effects. The reported side effects were consistent with the adverse effects in labeling for bupropion products. More than half of the patients who switched back to Wellbutrin XL 300 mg reported improvement of depression and/or abatement of side effects.
Given the temporal relationship between the switch to the generic product and the recurrence of depression and/or onset of side effects, these patients and physicians attributed these effects to poor performance of the generic product. These reported cases occurred at a time when sales data suggest that hundreds of thousands of patients using Wellbutrin XL were switched to the newly available Teva bupropion XL. The question is whether the reported lack of efficacy and/or new onset side effects in these patients who switched suggest a problem with the generic product, i.e., lack of bioequivalence to the branded product, or have some other explanation.
In order to evaluate this series of post-marketing reports, we have re-examined both the data on the bioequivalence of the two products (Wellbutrin XL and Teva's bupropion XL) and what is known about the natural history of treated depression.
What is the regulatory history of Wellbutrin and generic buproprion?
Bupropion hydrochloride is a drug used to treat Major Depressive Disorder (MDD). For many years bupropion was available only under the brand name Wellbutrin. It was first approved in 1985 as an immediate release (IR) tablet (Wellbutrin-IR) taken three times a day. In 1996, FDA approved a sustained-release tablet of bupropion (Wellbutrin SR), allowing twice a day dosing. In 2003, FDA approved an extended-release tablet of bupropion (Wellbutrin XL), allowing once a day dosing. Wellbutrin SR and Wellbutrin XL were approved based on the similarity of plasma levels of bupropion produced by these longer-acting products taken once or twice a day to the immediate-release product taken three times a day. The antidepressant effect of this drug does not appear for several weeks after initiation of treatment, and the effect is, in large part, related to long-acting metabolites. Therefore, no clinical effectiveness studies were considered necessary or required for the approval of Wellbutrin SR or Wellbutrin XL. Wellbutrin is owned by Smith Kline Beecham, a division of GlaxoSmithKline, and is manufactured by Biovail.
The law requires that generic drugs approved by FDA have the same active ingredient, dosage form, route of administration, and labeling as the branded product, and that the generic and branded drug be bioequivalent. The law also requires that generic drug applicants ensure the identity, quality, strength, and purity of their drug products. Bioequivalence means the generic drug's rate and extent of absorption do not show a significant difference from the branded drug's rate and extent of absorption. Statistics are used to analyze whether differences are considered significant. Generic drug products approved by FDA are therapeutically equivalent to the branded product. Therapeutically equivalent drugs generally may be substituted for each other with the expectation that the substituted product will produce the same clinical effect and safety profile when used according to the labeling.
In 2006, a generic XL version of bupropion, marketed as Budeprion XL, was approved by FDA. This generic formulation is manufactured by Impax Laboratories and distributed by Teva Pharmaceuticals. FDA approved this generic product based on evidence demonstrating bioequivalence between brand and generic bupropion XL (see below).
What was the basis for approval of Teva’s generic bupropion XL?
The basis for approval of Teva’s bupropion XL was that there was no significant difference in the rate and extent of absorption as measured by the plasma bupropion concentrations between 150 mg of the Teva XL product and 150 mg of Wellbutrin XL. Because of the potential risk of seizures at higher doses, the 300 mg strength was not studied. This practice is used when evaluating the pharmacokinetic profile of a drug in normal volunteers, especially when a drug’s adverse effects increase with dose. The pharmacokinetic profile is not expected to differ between 300 mg and 150 mg doses of bupropion.
The area under the drug plasma concentration over time curve (AUC) is a graphical and statistical representation of the total amount of drug absorbed. The average bupropion AUC from the volunteers receiving the Teva generic product under fasting conditions was 98% (90% CI, 91.9%-104.4%) of the average AUC from all of the same volunteers after receiving the Wellbutrin XL under fasting conditions. Under fed conditions the average bupropion AUC for the Teva product was 108% (90% CI 101.4% -115.4%) of that for the Wellbutrin XL product.
The average maximum bupropion plasma concentration (Cmax) produced by the Teva product was 89% (90% CI, 80.3% – 98.2%) of that produced in the same volunteers by Wellbutrin XL under fasting conditions and 110% (90% CI, 103.2% - 118.0%) under fed conditions.
The established bioequivalence limits provide that the entire 90% confidence interval (CI) for the generic/reference comparison of both AUC and Cmax be within 80% to 125%. Thus, the small differences observed in AUC and Cmax, with no consistent direction, are within the established limits for bioequivalence between brand name products and generic versions, and are not considered clinically relevant.
The major active metabolite, hydroxybupropion, which is responsible for much of bupropion’s effect, also met the AUC and Cmax bioequivalence limits.
The time to maximum drug plasma concentration (Tmax) was examined but is not required to be within any specified limits. The bupropion Tmax was faster for Teva’s XL product (2-3 hours) than Wellbutrin XL (5-6 hours). The median Tmax values for hydroxybuproprion (the active metabolite) with Teva’s product was 10 hours while Wellbutrin’s hydroxybuproprion Tmax was 12 hours (in both fasting and fed subjects). These differences in Tmax for both bupropion and its active metabolite, however, were not considered clinically significant. The somewhat more rapid times to maximum concentration, with no differences in the plasma bupropion concentrations (including the lowest levels, known as trough levels) throughout the day, would not lead to decreased effectiveness. This is supported by the fact that the bupropion Tmax of Teva’s XL product was similar to that of the marketed Wellbutrin SR and was, in fact, slower than that of Wellbutrin IR, a dosage form shown in clinical trials to be effective. The pharmacokinetic profiles of the generic and branded products do not support a conclusion that the reported lack of antidepressant effect and new onset side effects are the result of differences between the two products.
The figure below illustrates the concentration-time curves for the two formulations.
Mean plasma concentration of bupropion (Budeprion XL and Wellbutrin XL) as a function of time
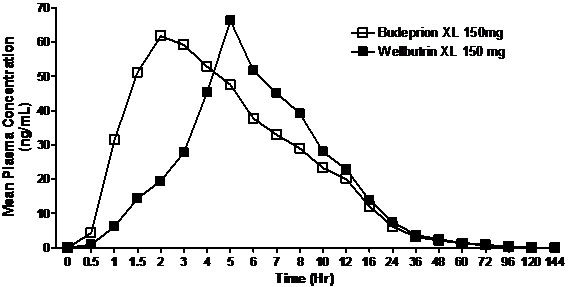
What other factors could account for the reported effects following a switch?
Natural history of depression
A factor that may account for the instances of recurring depression in the reported cases (see above) is the natural history of major depression, which can recur despite continued treatment, and has been shown to do so in controlled clinical studies. To demonstrate long-term effectiveness of antidepressants, patients who have responded to drug (e.g., bupropion) for several months are randomized to continuing the effective treatment or to a placebo (i.e., randomized withdrawal studies). In these studies, recurrence of depression is much more common in the placebo group, but also can return within weeks despite continued treatment with the active drug.1 One study of patients on duloxetine maintenance therapy, for example, found that approximately 5% of the patients on continued duloxetine had a recurrence of depression within four weeks.2 By six months, depression had recurred in approximately 20% of the patients on duloxetine. In a similar type of study of Wellbutrin SR, about 8% of patients receiving Wellbutrin SR had a recurrence of depression within four weeks.3 In both reports, recurrence of depression within two weeks after stopping active treatment was very rare. These rates of recurrence of depression in treated patients offer a fully satisfactory explanation for why some patients experience worsening of their depression following a switch to a generic product, just as some patients would have experienced recurrence without any such switch. Therefore, although it would seem reasonable to attribute worsened depression after a switch to a generic product to the change in treatment, for the reasons described, this conclusion would be incorrect.
In 2007, an average of approximately 1 million prescriptions per month were dispensed for all XL versions of bupropion.4 Of this number, an average of 40% of prescriptions per month were for Teva’s bupropion XL. It is unknown precisely how many of these generic prescriptions involved a switch from the branded product and how many represented an initiation of bupropion therapy.
The large number of prescriptions for this product, coupled with the data from the randomized withdrawal trials referenced above that have shown a recurrence rate for depression of approximately 5-8% during the first month of continued effective pharmacotherapy, suggests that there would be many thousands of people experiencing recurrence within 30 days of starting of therapy, regardless of whether patients are maintained on the branded product or switched to the generic product, both of which have been approved as safe and effective. To illustrate, if 10,000 persons switched from brand to generic in any given month with a recurrence rate of 5-8% per month, it would be expected that 500-800 of these individuals would experience worsening of their symptoms of depression during this timeframe, even with the drugs being equally effective. These calculations show that the reported cases of worsening of symptoms following a switch are far more likely to be a consequence of the natural course of treated MDD than of the small pharmacokinetic differences between the generic and branded product.
What is FDA's conclusion with respect to use of generic versions of Wellbutrin XL?
The FDA considers the generic form of bupropion XL 300 mg (Teva Pharmaceuticals) bioequivalent and therapeutically equivalent to (interchangeable with) Wellbutrin XL 300 mg. Although there are small differences in the pharmacokinetic profiles of these two formulations, they are not outside the established boundaries for equivalence nor are they different from other bupropion products known to be effective. The recurrent nature of MDD offers a scientifically reasonable explanation for the reports of lack of efficacy following a switch to a generic product. The adverse effects (e.g., headache, GI disorder, fatigue and anxiety) reported following a switch were relatively few in number and typical of adverse drug events reported in drug and placebo groups in most clinical trials (i.e., including, but not specifically for, bupropion). Although many of these adverse effects are seen soon after drug therapy is initiated, adverse effects are known to occur throughout the course of a patient’s therapy, as well as among patients on a stable dose of medicine or in patients receiving placebo. FDA continues to closely monitor reports of adverse events and therapeutic inequivalence.
References:
- Byrne SE, Rothschild AJ. Loss of antidepressant efficacy during maintenance therapy: possible mechanisms and treatments. J Clin Psychiatry. 1998;59(6):279-88.
- Perahia DG, Gilaberte I, Wang F, Wiltse CG, Huckins SA, Clemens JW, Montgomery
SA, Montejo AL, Detke MJ. Duloxetine in the prevention of relapse of major depressive disorder: double-blind placebo-controlled study. Br J Psychiatry. 2006;188:346-53.
- Weihs KL, Houser TL, Batey SR, Ascher JA, Bolden-Watson C, Donahue RM, Metz A. Continuation phase treatment with bupropion SR effectively decreases the risk for relapse of depression. Biol Psychiatry. 2002;51(9):753-61.
- Verispan, LLC. Vector One: National. Year 2007. Extracted 2-08.
 Back
to Top Back
to Top  Back to Bupropion Back to Bupropion
 PDF requires the free Adobe Acrobat Reader PDF requires the free Adobe Acrobat Reader
Date created: April 16, 2008
|
|



