 |
 |
 |
FDA
Home Page | Search FDA Site |
FDA A-Z Index | Contact FDA
![]()
This Plan outlines key performance goals and strategies designed to achieve these goals during FY 1999. The Plan serves several purposes:
The Plan is organized according to the six objectives outlined in Section 406(b) of FDAMA.
These objectives address critical components of FDA's responsibilities. The Agency, working in collaboration with key players in both the public and private sector, will pursue each objective as part of a total consumer health protection and enhancement system. The process begins with the research and development of new products with great health- and life-sustaining potential, and ends with the safe and effective consumption of these products. Figure 5 illustrates how FDAMA objectives are crucial elements of FDA's total contribution to beneficial public health outcomes.
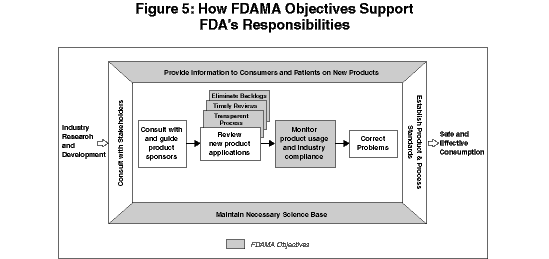
The six 406(b) objectives are addressed in five sections below. The five sections examine the FDAMA objectives in order by objective (A, B, C, D, and E&F). Each section provides:
| Identification of Needs | Outlines the unmet demands stated by law and expressed by the Agency's stakeholders, which FDA must address to achieve the FDAMA objective and to fulfill its mission. |
| Stakeholder Views | Selected stakeholder opinions on the importance of the need being addressed. |
| Current Innovations and Reinventions | Creative improvements FDA has underway that will help achieve objectives. |
| Plan for Meeting Statutory Requirements and Public Expectations | Key strategies that are planned for the future that will narrow the gap between expectations and current capabilities. |
| Performance Goals for FY 1999 | FY 1999 goals are based on final Congressional appropriations and may be subject to adjustment pending Agency resource allocation decisions. |
Maximizing the availability and clarity of information about the process for review of applications and submissions (including petitions, notifications and any other similar forms of requests) made under this Act.
FDA's ability to provide clear, adequate, and timely information on its application reviewprocesses must be improved by making FDA processes transparent to stakeholders and involving stakeholders early in the review process.
Make FDA Processes Transparent
While the Agency has developed written information (i.e., regulations, guidance documents, or internal procedures) on its review processes and requirements, more needs to be done to ensure that stakeholders understand FDA requirements. This lack of understanding is reflected in the quality of regulatory submissions received by FDA. Transparent processes also include openness on how FDA develops its requirements and how those requirements are applied within the Agency during the review process.
Collaborate with Stakeholders Early in the Regulatory Decisionmaking Processes
In passing FDAMA, the Congress expected major improvements on how products are reviewed and approved by FDA. To meet this expectation, FDA must change how it responds to the product applicants during the review process--from being reactive to proactive through early applicant consultations. By consultation with product sponsors, the Agency will be able to help define the critical issues that must be addressed in a product application, to define the types of clinical trials that appear necessary, and to avoid unnecessary effort. This shifting of resources is not, however, without cost, and additional resources will be needed to meet the increasing number of product submissions generated by the doubling of biomedical research funding at the National Institutes of Health and by the regulated industry.
Stakeholders endorsed the concept of a more open and collaborative relationship between FDA and its regulatory colleagues and industry. Many stakeholders commended FDA for the efforts the Agency has already made to address this objective. Requests for improved communication about application review processes emphasized not only communication from FDA to industry, but also greater stakeholder participation in regulatory decisionmaking. The examples below illustrate some of the further improvements stakeholders requested:
FDA is improving its review processes and specific product applications through collaborative agreements, process re-engineering, and information technology.
Agreements Among FDA, Industry, and Others Enhance Review Processes
FDA, academia, and industry are working to establish a program to provide research to inform and assist FDA in developing regulations and guidance regarding the types of product quality information that should be submitted in a product application (e.g., Collaboration for Drug Development Improvement and Product Quality Research Initiative).
FDA collaborates with regulatory authorities of Europe and Japan on drug development requirements (e.g., International Harmonization).
FDA Continues To Improve Review Processes Through Process Re-engineering.
FDA's medical device program improved by providing manufacturers with regulatory options to reduce regulatory burden for lower risk products and by improving communication with manufacturers. As part of the Reinventing Government Initiative (REGO), FDA has simplified the filing process by consolidating review application forms for biotechnology-based drugs, blood, vaccines, and other drugs into just one form. This enables companies to provide higher quality submissions to the FDA and reduces their application preparation time.
During FY 1997 and early FY 1998, the Foods Program conducted under contract a review of deficiencies in over 600 industry-submitted food and color additive petitions. CFSAN currently is reviewing the contractor's report and expects to use the information to improve guidance to petitioners and to implement a stronger refusal to file policy.
FDA Uses Information Technology To Improve Access of Review Processes
The FDA website (www.fda.gov) provides specific information to particular stakeholder groups: consumer, industry, state and local officials, patients, health professionals, women, and children.
FDA has published information on its review processes to assist applicants. For example, the FDA Center for Drug Evaluation and Research (CDER) Handbook is available on the Internet.
The Foods Program is completing testing on a document management and workflow system that will replace the current tracking system for petition reviews and will make petition data available on demand in electronic format on reviewer's and administrator's desktops. The new workflow tracking system will permit realtime access to detailed information on petition status and tasks.
Section 903 of the FD&C Act, as amended by FDAMA, authorizes the Commissioner to conduct educational and public information programs relating to the responsibilities of FDA. Under FDAMA (Section 406), FDA's mission is expanded to include the prompt review of clinical research and regulatory submissions, harmonization of regulatory requirements with other countries, and consultation of various experts in fulfilling the mission.
FDA's plan for meeting these statutory requirements will encompass a variety of actions intended to make Agency processes transparent and to improve collaboration between product sponsors and the Agency. These include:
The table provided in this section links the performance goals and measures with statutory requirements addressing information about the review processes. Under the FD&C Act, the Commissioner is authorized to conduct educational and public information programs relating to FDA's responsibilities. These performance goals illustrate two types of efforts. The first type identifies the development of a method that can be applied to a review process. An example would be to recognize a standard used for a medical device review. The second type identifies an improvement to enhance the Agency's ability to provide updated information or to achieve greater capability and capacity for accepting electronic regulatory submissions.
Highlighted below are key performance goals for FY 1999 in the area of electronic regulatory submissions. These goals are critical to the Agency's ability to provide timely review of clinical research and regulatory submissions, which is the intent of FDAMA. For more complete identification of performance goals and statutory requirements see the table at the end of this section.
Complete the development of industry guidance required for electronic submission by the end of FY 2002.
Achieve electronic submission capability for certificates to foreign governments.
Achieve capability and capacity for electronic submission and archiving of information required to submit New Drug Applications (NDAs) without paper copy by the end of FY 2002.
Achieve capability and capacity for electronic submission and archiving of Abbreviated New Drug Applications (ANDAs) by the end of FY 2002.
| Statutory Authority | Relevant Statute and/or Regulation | Relevant FY 1999 Performance Goals | FY 1997 Performance Baseline |
|---|---|---|---|
| Applicants are invited to meet with FDA before submitting an application to discuss the presentation and format of supporting information. If the applicant and FDA agree, the applicant may submit tabulations of patient data and case report forms in a form other than hard copy, for example, on microfiche or computer tapes. | FD&C Act, Section 505 and 21 Code of Federal Regulations (CFR) 314.50 (f)(4) | By the end of FY 2002, CDER will complete development of industry guidance required for electronic submission. | In FY 1997, electronic signature guidance was published. |
| Before 30 days after the date of submission of an application to export a drug, the FDA must review the application to determine if it meets all applicable requirements. | FD&C Act, Section 801(e) and 802, 21 CFR 210, Drug Export Amendments Act of 1986 (PL. 99-660), FDA Export Reform & Enhancement Act of 1996 | By the end of FY 1999, CDER will achieve electronic submission capability for certificates to foreign governments | In FY 1998, develop and pilot Export Certificate Program. |
| For records submitted to the Agency, persons may use electronic records in lieu of paper records or electronic signatures in lieu of traditional signatures, in whole or in part, provided that certain requirements are met. | FD&C Act, Sections 201 - 903; PHS Act, Section 3512, 21 CFR 11 | By the end of FY 2002, CDER will achieve capability and capacity for electronic submission and archiving of information required to submit NDAs without paper copy.By the end of FY 2002, CDER will achieve capability and capacity for electronic submission and archiving of ANDAs. | By FY 1997, establish the structure of the Electronic Document Room (EDR). By FY 1997, establish the structure of EDR. |
| Any record of the FDA that is disclosed in an authorized manner to any member of the public is available for disclosure to all members of the public, except that data and information subject to the exemptions established in 21 CFR 20.61 for trade secrets and confidential commercial or financial information, and in Section 20.63 for person privacy, shall be disclosed only to the persons for the protection of whom these exemptions exist. | FD&C Act, Sections 201-903, 5 United States Code 552, 21 CFR 20 | By the end of FY 2002, CDER will make publicly releasable information available via Internet. | By FY 1998, the Electronic Document Room, as required by the Electronic Freedom of Information Act, will be initiated. |
| Publish regulations for adequate and well-controlled clinical trials by 4/9/98 and substantial evidence by 10/9/98 | Animal Drug Availability Act (ADAA), (P.L. 104-250) Section 2(e) | FDA Center for Veterinary Medicine (CVM) will revise Investigational New Animal Drug Application procedural regulations and implement provisions of the ADAA and CVM's REGO initiatives. | ADAA enacted by 10/9/96 |
| Recognize and approve list of standards suitable for use in application review. | FD&C Act, Sections 514(b) and (c) | FDA Center for Devices and Radiologic Health (CDRH) will recognize over 415 standards for use in application review and update the list of recognized standards. | 0 recognized |
FY 2000 Performance Goals are not identified in this Plan. Specification of these goals is dependent upon final determination of the President's FY 2000 Budget submission to Congress.
Maximize the availability and clarity of information for consumers and patients concerning new products.
FDA is reviewing applications for new drugs, biologics, medical devices and food additives more quickly. Dissemination of information that will enhance consumption decisions about these new products must keep pace with the products' earlier availability. The Agency would like to provide timely information to consumers and patients, however, in some instances products are reaching the market faster than FDA can inform its stakeholders. The Agency's ability to disseminate information must be enhanced by upgrading its technology, its computers, and the training of its employees to keep abreast with the latest developments in technology. FDA is under pressure from Congress, the medical community, patients, and industry to provide timely unbiased information to its stakeholders.
Stakeholders strongly agree that maximizing the availability and clarity of information to consumers and patients about new FDA-regulated products is a priority. A selection of stakeholder comments is provided below:
FDA is currently expanding its information for consumers and patients. The following are illustrations of the information exchange:
Collaboration
The Agency is collaborating with industry to inform patients and consumers of the availability of new drugs (prescription and over-the-counter [OTC] drugs). FDA engages in cooperative research with industry for new food items as well as collaborates with industry to bring better food labels and information to its stakeholders.
The Agency is collaborating with industry to provide technical, non-financial assistance to manufacturers to enable them to bring their products that meet FDA standards to the market more quickly.
Outreach
FDA has an outreach program to keep physicians informed of new drugs available to their patients. The Agency is working cooperatively with the drug industry, consumers, and patients to inform them of new drugs and emerging new drugs. Patients are able to receive information on new therapies approved by foreign countries before they are approved by the Agency. Additionally, the Agency's Public Affairs Specialists in the field offices furnish information to interested consumers and patients concerning new drugs, devices, etc.
FDA delivers educational and technical assistance in the area of food safety messages and uses. The FDA Consumer/Fact Sheets and National Food Safety Hotlines are part of the Agency's outreach. The Internet is used to bring new information to consumers and patients. Each Center has its own web page. Many of these pages are interactive and allow the user to communicate with the Agency directly. Printed materials are provided to those that are without Internet capabilities, and many of the materials are in several languages. These materials help to inform consumers and patient about new drugs. The Veterinary Newsletter, exhibits, and Public Affairs Specialists programs keep the veterinary community abreast of the newest drugs and technology being developed.
During the 20th century, the nation has witnessed a more dramatic extension of longevity than humankind has ever seen. The Agency is making a concerted effort to ensure that older persons, their families, and their communities are aware of FDA's responsibilities and how the Agency can be a resource for them in improving the quality of their lives.
FDA's consumer protection and public health mission plays a particularly important role in building a sound health foundation for ensuring quality of a long life for older persons. The needs of the U.S. aging population are stimulating innovative research and technological advancements for both preventing and treating disease. The Agency makes a meaningful contribution to this research by facilitating the timely availability of safe and effective products, keeping unsafe or ineffective products off the market, and providing easily understandable and meaningful information about the availability of new products, as well as how to use products safely and effectively. In October 1998, the United Nations launched the International Year of the Older Person 1999 to bring global attention to the phenomenon of an aging world and the need to begin to establish the policies, programs, and services needed to meet the needs of an aging world. The Agency is an active participant in this initiative.
Section 406(b) requires the Agency to maximize the availability and clarity of information for consumers and patients concerning new products. FDA is engaged in a variety of activities to fulfill this requirement that revolve around four themes. First are Agency efforts to ensure that product information is tailored to meet the special needs of diverse populations. One example is the implementation of public awareness campaigns for consumers, i.e., Take Time to Care, Office of Women's Health; Mammography Awareness Seminars; Food Safety Programs (Fight BAC!); Over the Counter Labeling Changes (OTC) Campaign; and the Partnership for Food Safety Education. As the population becomes more culturally diverse, FDA must reach out to consumers in ways they will understand. For instance, Public Affairs Specialists give seminars on new drug therapies, health fraud, labeling, etc. in different languages to fulfill the needs of diverse populations.
The Agency is entering into an increasing number of stakeholder "collaborations" to achieve a multiplier effect ( e.g., with print media, radio, television, industry, other federal agencies, consumers, health professionals, and associations). Another example is implementation of the Pharmacist Education Outreach Program to assist pharmacists in explaining the drug approval process to consumers.
Another approach is focusing FDA resources so that patients are an integral part of the health care decisionmaking process. FDA has established programs to make promising investigational drugs, therapies, and devices available to patients with serious and life-threatening conditions. For example, FDA has also included patient representatives on advisory committees considering products for HIV/AIDS, cancer, and other serious diseases.
The technological revolution provides the Agency the tools to offer quick access to a wide range of information to consumers through various methods. The Internet is being used as a means for two-way communication--both to disseminate information about new products and to quickly answer questions about new and existing products. Additionally, the Agency will participate with NIH in the establishment of (under Section 402 of the Public Health Service Act) a registry of publicly and privately funded clinical trials for experimental drugs and biologics being tested for serious or life-threatening medical conditions. This registry will simplify the process of obtaining information.
The table provided in this section links the performance goals and the measures with statutory requirements to regulate information provided to consumers and to ensure that consumers understand OTC drug information. The FY 1999 performance goals focus on both OTC and prescription drugs. FDA wants consumers and patients to receive and to be able to refer to the highest quality information when taking either OTC or prescription medications.
Highlighted below are key performance goals for FY 1999. These goals seek to provide drug information, in easily understood language, to consumers and patients faster through various outreach efforts. For more complete identification of performance goals and statutory requirements see the table at the end of this section.
Evaluate drug information provided to 75 percent of individuals receiving new prescriptions.
Improve OTC information and consumers' ability to understand it by 2001.
| Statutory Authority | Relevant Statute and/or Regulation | Relevant FY 1999 Performance Goals | FY 1997 Performance Baseline | FY 1998 Performance Baseline |
|---|---|---|---|---|
| FDA regulates prescription drug advertising and labeling by monitoring all prescription drug promotions, enforcing the laws and regulations, developing new guidance, and conducting research to support the program. | FD&C Act Sections 502(n) and 505 and 21 CFR 200-202 | FDA will a) evaluate the availability, quality, and usefulness of prescription drug information provided to 75 percent of individuals receiving new prescriptions; and b) complete two studies that will aid in development of comprehensive drug information. | In 1996, 65 percent of patients received written information about prescription drugs. Assessments are underway to determine the degree to which this information meets the criteria for "useful" information. | Initiate partnerships with three major organizations |
| Target 25 percent of review documents processed using Electronic Data Management System (EDMS) | ||||
| FDA is responsible for assuring that OTC drugs are safe and effective for use--this includes improving the legibility and clarity of all OTC drug labels as well as consumer's ability to comprehend important warnings and usage directions. | FD&C Act Section 502 and 21 CFR 201, 21 CFR 211.132 | By the end of FY 2001, CDER will improve the legibility and clarity of OTC drug labels, improve the consumer's ability to read and understand important warnings and usage directions, and complete two studies that will aid in development of comprehensive drug information. | Federal Register publication on February 27, 1997 (62 FR 9024) published a proposal providing for standardized format for labeling. Study topics have been identified and studies are being designated. |
FY 2000 Performance Goals are not identified in this Plan. Specification of these goals is dependent upon final determination of the President's FY 2000 Budget submission to Congress.
![]()