|
 |
 |
|
 |
FDA Home Page | CBER A-Z Index | CBER Search | Contact CBER | CBER Home Page |
||||
This guidance is one of three guidances intended to assist developers of medical imaging drug and biological products (medical imaging agents) in planning and coordinating their clinical investigations and preparing and submitting investigational new drug applications (INDs), new drug applications (NDAs), biologics license applications (BLAs), abbreviated NDAs (ANDAs), and supplements to NDAs or BLAs. The three guidances are: Part 1: Conducting Safety Assessments; Part 2: Clinical Indications; and Part 3: Design, Analysis, and Interpretation of Clinical Studies. Medical imaging agents generally are governed by the same regulations as other drugs or biological products. However, because medical imaging agents are used solely to diagnose and monitor diseases or conditions as opposed to treat them, development programs for medical imaging agents can be tailored to reflect these particular uses. Specifically, this guidance discusses our recommendations on selecting and studying clinical indications for medical imaging agents administered in vivo.2 FDA's guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidances describe the Agency's current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidances means that something is suggested or recommended, but not required. A glossary of common terms used in diagnostic medical imaging is provided at the end of this document.
This guidance discusses medical imaging agents that are administered in vivo and are used for diagnosis or monitoring with a variety of modalities, such as radiography, computed tomography (CT), ultrasonography, magnetic resonance imaging (MRI), and radionuclide imaging. The guidance is not intended to apply to the development of in vitro diagnostic or therapeutic uses of these agents.3 Medical imaging agents can be classified into at least two general categories, contrast agents and diagnostic radiopharmaceuticals.
As used in this guidance, a contrast agent is a medical imaging agent used to improve the visualization of tissues, organs, and physiologic processes by increasing the relative difference of imaging signal intensities in adjacent regions of the body. Products include, but are not limited to (1) iodinated compounds used in radiography and CT; (2) paramagnetic metallic ions (such as ions of gadolinium, iron, and manganese) linked to a variety of molecules and used in MRI; and (3) microbubbles, microaerosomes, and related microparticles used in diagnostic ultrasonography.
As used in this guidance, a diagnostic radiopharmaceutical is (1) an article that is intended for use in the diagnosis or monitoring of a disease or a manifestation of a disease in humans and that exhibits spontaneous disintegration of unstable nuclei with the emission of nuclear particles or photons or (2) any nonradioactive reagent kit or nuclide generator that is intended to be used in the preparation of such an article.4 The FDA interprets this definition to include articles that exhibit spontaneous disintegration leading to the reconstruction of unstable nuclei and the subsequent emission of nuclear particles or photons. Diagnostic radiopharmaceuticals are generally radioactive drugs or biological products that contain a radionuclide that typically is linked to a ligand or carrier.5 These products are used with planar imaging, single photon emission computed tomography (SPECT), positron emission tomography (PET), or with other radiation detection probes. Diagnostic radiopharmaceuticals used for imaging typically have two distinct components.
As technology advances, new products may emerge that do not fit into the traditional categories of contrast agents and radiopharmaceuticals (e.g., agents for optical imaging, magnetic resonance spectroscopy, combined contrast and functional imaging). It is anticipated, however, that the general principles discussed here could apply to these new diagnostic products. Developers of these products should contact the appropriate reviewing division for advice on product development.
The labeled indications for medical imaging agents fall within the following general categories:
The above categories do not represent a hierarchy or progression (e.g., a structure delineation indication does not need to precede a disease assessment indication). In addition, indications from different categories could be granted for the same imaging agent. Approval also may be possible for categories of indications not listed above. Under section 505(d) of the Federal Food, Drug, and Cosmetic Act (the Act) (21 U.S.C. 355(d)), FDA cannot approve a new drug application (NDA) unless it contains adequate tests demonstrating whether the proposed drug product is safe for use under the conditions prescribed, recommended, or suggested in its proposed labeling.6 All drugs have risks, including risks related to the intrinsic properties of the drug, the administration process, the reactions of the patient, and incorrect diagnostic information. Incorrect diagnostic information includes inaccurate structural, functional, physiological, or biochemical information; false positive or false negative diagnostic determinations; and information leading to inappropriate decisions in diagnostic or therapeutic management. Even if risks are found to be small, all drug development programs must obtain evidence of drug effectiveness under section 505 of the Act. Simply generating an image, for which the implications to the patient are not understood, does not confer benefits to the patient. In determining the most appropriate indication for a medical imaging agent, special considerations may apply to agents that may pose significant patient risk, for example, biological medical imaging agents that are frequently immunogenic. The development of antibodies after intermittent, repeated administration can alter the pharmacokinetics, biodistribution, safety, and/or imaging properties of such agents and, potentially, of immunologically related agents. For agents that pose significant risk and where the clinical benefit is generally not readily apparent, an indication of disease or pathology detection or assessment or diagnostic or therapeutic patient management is more appropriate. If one of the other indications (i.e., structure delineation or functional, physical or biochemical assessment) will be sought for an agent that may pose significant patient risk, we recommend that the development plan be discussed with the review division.
As described in the following sub-sections, at least two types of labeled indications for structure delineation are possible: (1) locating and outlining normal (or variants of normal) anatomic structures and (2) distinguishing between normal and abnormal anatomy in a defined clinical setting. Ordinarily, the ability to locate and outline normal structures or distinguish between normal and abnormal anatomy can speak for itself with respect to the clinical value of the information and will not require additional information substantiating clinical usefulness.
We recommend that a medical imaging agent intended for disease or pathology detection be able to detect and locate a specific disease or pathological state in at least one defined clinical setting.7 The medical imaging agent could be used alone or in combination with other diagnostic procedures to achieve this labeled indication. Examples of medical imaging agents for which this type of indication may be appropriate include:
We recommend that efficacy trials for these indications be conducted in subjects presenting for diagnostic evaluation of a specific disease or condition in a defined clinical setting. This is because the likelihood of disease or the spectrum of disease (e.g., severity or stage) is dependent on the clinical setting. Examples of two common clinical settings are (1) providing a diagnosis in patients with suspected disease and (2) monitoring and assessing the extent, rate of progression, or other aspects of the specific disease in patients previously diagnosed with the disease. An indication of detection of disease or pathology in an asymptomatic population (a screening indication) may be appropriate if the sensitivity of the imaging modality is high enough and the rate of false positives is low enough (see also diagnostic or therapeutic patient management, section III.D). It is likely that the clinical usefulness and the diagnostic performance of the medical imaging agent will differ in each clinical setting.8 We recommend that if a medical imaging agent is being developed to diagnose a particular disease, efficacy trials generally enroll subjects in whom the disease status is unknown, but in whom specific aspects of the clinical presentation have led to the desire for more diagnostic information. That is, we recommend that the trials include the intended population in the appropriate clinical setting. Data from subjects known definitely to have (or to not have) the disease of interest may be of limited value because estimates of diagnostic performance derived from a known disease population may not apply to performance in the intended population.
We recommend that a medical imaging agent intended to provide functional, physiological, or biochemical assessment be able to evaluate the function, physiology, or biochemistry of a tissue, organ system, or body region. This type of indication could apply to agents used to detect either a reduction or an increase of a normal functional, physiological, or biochemical process. The indication functional, physiological, or biochemical assessment could be limited to assessment of functional, physiological, or biochemical processes when disturbances of these processes are common to several diseases or conditions and they are not diagnostic for any particular disease or condition. The indication functional, physiological, or biochemical assessment is appropriate for patients when evaluations of functional, physiological, or biochemical aspects of a tissue, organ, or body region would provide clinically useful information. Examples of medical imaging agents with functional, physiological, or biochemical assessment indications include:
To establish efficacy in clinical studies, we recommend that the functional, physiological, or biochemical measurements of the medical imaging agent be compared with those of a reference product or a procedure of known high validity (i.e., a truth standard). Ideally, we recommend that the high validity of this reference product or truth standard be documented thoroughly and critically before its use in clinical studies intended to demonstrate effectiveness of the test-imaging agent. We recommend that a functional indication be studied in the wide spectrum of diseases and disease severity states that affect the functional endpoint. For example, a sponsor might seek an indication of measuring myocardial left ventricular function. To ensure that the test is valid in the patient population most likely to be referred for testing, the sponsor might design studies that include subjects with different cardiac diseases, such as dilated cardiomyopathy, valvulopathy, hypertrophic cardiomyopathy, and myocardial infarction, including subjects with normal function as well as those with mild, moderate, and severe dysfunction. In that case, separate studies for each disease would not be needed. If no standard of truth applies to the proposed use of a medical imaging agent for functional, physiological, or biochemical assessment, we recommend that a clinical trial be conducted to determine that the findings are clinically useful (see section IV.B).
We recommend that a medical imaging agent intended for the indication diagnostic or therapeutic patient management be able to improve patient management decisions (e.g., the need for further diagnostic testing or the use of specific therapeutic interventions) or improve patient outcomes when used in a defined clinical setting.9 Included in this indication is the ability to provide information (such as the presence of a certain receptor in a type of cancer patient) that can predict survival or patient response to a particular type of therapeutic drug. To obtain approval for a diagnostic or therapeutic patient management indication, we recommend that adequate and well-controlled investigations demonstrate that patient management decisions or outcomes are, in fact, improved by use of the medical imaging agent. The medical imaging agent can be used alone or in combination with other diagnostic procedures to achieve this labeled indication. Studies might involve randomization into one arm with testing and patient management based on the testing results in accordance with a prespecified algorithm compared to a nontesting arm that proceeded to treatment as defined by current standards. Patient outcomes such as recovery, survival, and response rates that are based on the treatment standard could be collected and compared. Examples of medical imaging agents for which this type of indication may be appropriate are:
We recommend that the trials demonstrate that diagnostic or therapeutic management is improved when using the medical imaging agent compared to management without use of the medical imaging agent. The medical imaging agent can be used in conjunction with other tests to influence a patient diagnostic or therapeutic management decision. We suggest that it would not be sufficient simply to demonstrate that the results of the test drug were used to direct a change in patient management, even to an intervention that is well established. Rather, we recommend that the sponsor establish whether the change was better or worse for the patient. For example, when using a new imaging agent in determining whether to perform breast biopsy versus repeat clinical breast examination followed by mammography, the sponsor should show whether use of the test drug results in fewer or greater numbers of unnecessary biopsies or undiagnosed cancers than use of a comparator. We recommend that this principle also be applied to studies to demonstrate a therapeutic patient management claim: the sponsor should show that use of the new imaging agent leads to better patient therapy choices than result from the use of existing methods of managing therapy. If sponsors do not wish to perform such follow up, we recommend that they instead seek an indication for disease or pathology detection or assessment. To obtain the indication diagnostic or therapeutic patient management for a medical imaging agent that identifies unrecognized disease in asymptomatic individuals (e.g., used in a screening setting), we recommend that a sponsor show that use of the test decreases morbidity or mortality, or provide existing data that show that early detection and treatment of the disease decreases morbidity or mortality.
The indication categories outlined above are flexible, and indications for medical imaging agents need not be mutually exclusive. A labeled indication can include several indication categories. For example, a diagnostic radiopharmaceutical could be developed as an aid in the diagnosis of lung cancer for the labeled indication disease or pathology detection or assessment. This diagnostic radiopharmaceutical could also be evaluated in subpopulations of patients with lung cancer for its ability to provide information that leads directly to appropriate therapeutic management decisions (e.g., using test results to determine what combination of surgery, radiotherapy, and chemotherapy is most appropriate). Structural and functional aspects of diseases or conditions sometimes are evaluated together with imaging in clinical practice (e.g., use of a contrast agent to evaluate cardiac anatomy and segmental wall motion). In such cases, we recommend that clinical studies evaluate the effect of the imaging agent on assessments of both structure and function. Functional evaluations of diseases or conditions may be accomplished for various purposes. For example, a drug may have a functional indication for the evaluation of cardiac ejection fraction. Subsequently, the drug may be developed for a therapeutic management indication for the evaluation of perfusion or wall motion abnormalities to predict response to surgical intervention. For indications that do not fall within the categories identified above (e.g., providing prognostic information based on imaged gene expression), we recommend that the applicant or sponsor consult FDA on the nature of the desired labeled indication and how to establish effectiveness for it.
In general, establishing effectiveness has two components: (1) establishing the accuracy of the test and (2) establishing the clinical value of the test. In some cases, a test that provides accurate information in describing a clinical condition is of well-established value. Generally, this is true for proposed indications for structure delineation and disease or pathology detection or assessment. When there are established methods of seeking similar information and the only issue is comparing the accuracy of the new and old methods, the clinical usefulness of the indication need not be reestablished. Many functional, physiological, or biochemical assessments are similarly well established as useful (ejection fraction, renal function, myocardial wall motion) but others (glucose utilization by various parts of the body, presence of serotonin receptors, cerebral blood flow, palmitate metabolism) may not be. Where the clinical value of valid information is not established, we recommend that additional information establishing its value be developed. This recommendation applies to all drugs, including therapeutic drugs, for which the indication or mechanism of action for an indication is not accepted or well understood in the medical community. Demonstration of improved patient management means more than assessment of the accuracy of the test. Either by reference to prior data or through new trials, we recommend that this claim show that the test really makes a difference in outcome or management. Of course, the impact of a test may be considered obvious (e.g., staging of breast cancer is disease or pathology detection or assessment indication). In some cases, the test will have plain therapeutic implications, as would be the case for effective staging of some other malignancies, although, in many cases, trial data should be collected. We recommend that investigations establish the validity (generally assessed by describing the sensitivity, specificity, positive predictable value and negative predictive value in relevant settings) and reliability (how reproducible the test results are) of the imaging agent. These test characteristics can provide information on risk-benefit as well, including estimates of risk of incorrect diagnosis. Safety information obtained in studies (see the companion guidance Part 3: Design, Analysis, and Interpretation of Clinical Studies) will also contribute to an Agency risk-benefit assessment. The clinical usefulness of an imaging agent may be obvious from a description of what it can demonstrate (or supportable by evaluation of the literature), or it may be appropriate to demonstrate the agent's usefulness. We recommend that clinical studies and related methods for establishing effectiveness be performed in defined clinical settings that reflect the proposed indications.
To establish efficacy in clinical studies, we recommend that the accuracy and/or validity of the structural delineation, functional, physiological, or biochemical assessment and disease or pathology detection generally be demonstrated by comparing the performance of the medical imaging agent with that of a reference product or a truth standard in a relevant clinical setting. To provide adequate estimates of the validity and reliability of the medical imaging agent over the full range of conditions for which it is intended to be used, we recommend that medical imaging agents be evaluated in studies with appropriate representation of sufficient numbers of subjects (1) with and without the abnormalities or diseases in question (over the full spectrum of the condition or disease presentation) and (2) with other conditions, processes, or diseases that could affect the interpretation of the imaging results (e.g., inflammation, neoplasm, infection, trauma). We recommend that sponsors justify the inclusion or exclusion of selected subpopulations during clinical development. We recommend that studies of agents for functional, physiological, or biochemical assessment indications provide a quantitative or qualitative understanding of how the measurement varies in normal and abnormal subjects or tissues, including the variable's normal range, distribution, and confidence intervals in these subjects or tissues. We believe it is critical to identify the range that is normal and the values that indicate an abnormality. When possible, we recommend that the minimum detectable limits and reproducibility of the measurement be assessed. Reproducibility assessments are most meaningful when performed within the same subject. However, under some circumstances this practice might be unethical, in which case the sponsor should consider alternative approaches to testing reproducibility (e.g., in animals). In cases when a valid reference product or a truth standard is unavailable or infeasible, the validity of the information obtained can be demonstrated in clinical studies showing a beneficial clinical outcome. We recommend that the sponsor discuss these issues with the Agency prior to initiation of phase 3 studies.
Under section 505 of the Act (or, for a biologic, section 351 of the PHS Act) and its implementing regulations, FDA cannot approve a drug without evidence that the drug's benefit to patients outweighs its risks:
The use of medical imaging agents without defined benefits and without an understanding of how the imaging results can be used for patient management might cause harm to patients even if the agent has low toxicity. Such harm might include (1) conducting unnecessary diagnostic testing based on the results of the agent, (2) directing patients to invasive procedures or inappropriate or unnecessary therapy, and (3) creating unnecessary patient anxiety from abnormal test results. Medical imaging results may have clinical usefulness in some settings but not in others; it is, therefore, important to prospectively define and study the imaging agent in the clinical setting of intended use. We recommend that a medical imaging agent be able to provide accurate and reliable information that, in one of a number of ways, facilitates clinical management, including (1) helping make an accurate diagnosis, (2) contributing to beneficial clinical outcome (e.g., by helping choose the right therapy), or (3) providing accurate prognostic information. All indications under section III should reflect these benefits, which are then weighed by FDA against the agent's risks as part of an approval decision. Once clinical usefulness is established, other benefits of imaging agents, such as safety advantages and enhanced convenience to patients over existing marketed products, can be considered. Depending on the specific indication, clinical usefulness can generally be established in two ways: (1) by direct demonstration in studies carried out during clinical development and (2) by reference to historical data. In circumstances when the measure is well established as useful in the medical literature, the clinical benefit of the measure does not need to be re-established (e.g., ejection fraction or myocardial wall motion are widely used measures of cardiac function with known prognostic and therapeutic implications). Even if the new measure has not been used before, we suggest that clinical usefulness can be established historically when the information being obtained has been shown to be useful when obtained by other means. For example, if a product is able to establish the early detection of colon polyps without the need for colonoscopy, the clinical benefit of the use of this product can be inferred because treatment is available for this disease (polypectomy), and the test would allow people to avoid unnecessary colonoscopy (i.e., clinical usefulness has been established indirectly). In such situations, clinical usefulness can be documented by a critical and thorough analysis of the medical literature and any historical precedents. For indications in which it cannot be established from prior knowledge, we recommend that clinical usefulness be established through new trials during development. For example, we recommend that clinical usefulness be established directly for a medical imaging agent that has been shown in a research setting to bind specifically to particular receptors, but where it has not yet been established that assessment of such binding adds to the accuracy of diagnosis, contributes to beneficial clinical outcome, or provides accurate prognostic information. For novel technologies relying on mechanisms for imaging never approved before, we recommend that a plan for establishing clinical usefulness be incorporated into the development plan of a medical imaging agent. In general, we recommend that clinical usefulness be evaluated prospectively in the principal clinical studies of efficacy. We recommend that sponsors assess how the novel technology imaging results are used and how usefulness to the patient is confirmed. For a contrast agent to be considered clinically useful, we recommend that, when used in combination with an imaging device, the agent be able to provide useful information or other advantages (such as improved imaging time or convenience) beyond that obtained by the imaging device alone. That is, we recommend that imaging with the contrast agent have added benefit when compared to imaging without the contrast agent. To illustrate how effectiveness could be evaluated, consider the following possible approaches:
Note: Situation 2 would be similar to imaging with and without the new test to determine the contribution of the new imaging test. For 1 and 2, the results of the new test would be presumed to be of prognostic, therapeutic, or diagnostic value, and the new imaging drug would be presumed to improve these aspects. If that is not the case, we recommend that the value be documented through a randomized clinical trial. The new test (or the new test added to the standard testing battery) could be compared to standard testing without the new test to determine if the new test improves clinical outcomes or prognosis. Refer to Part 3 of the medical imaging guidances, section IV.D.1, for additional discussion.
We recommend that a defined clinical setting reflect the circumstances and conditions under which the medical imaging agent is intended to be used.10 Generally, the choice of anticipated labeled indications will determine the clinical setting for the trials. In some cases, an appropriately designed trial may contain several clinical settings. For example, a medical imaging agent intended to detect prostate cancer (a disease specific indication) could be developed for use in different defined clinical settings such as:
We recommend that the circumstances and conditions under which the medical imaging agent is intended for use be evaluated in clinical trials and be described in the labeling using the following mechanisms:
Pooling efficacy data (additive derivation of summary statistics) across defined clinical settings may only be of limited value because differences in disease prevalence and in pathophysiology may result in different diagnostic performance (sensitivity, specificity, positive and negative predictive value) in different settings. Pooled results may suggest that the product is useful in all the evaluated clinical settings and may obscure the evidence of differential usefulness in each one of the settings. Of course, data from independent trials in different clinical settings may be useful in determining the overall labeling in one or more clinical settings. The number and type of populations to be studied depends on the type of the indication and clinical uses sought by the sponsor. If there are data showing that the benefits from use of a medical imaging agent in a particular clinical setting exceed the risks, that can be reflected on the labeling.
On occasion, the medical literature may provide critical information on various aspects of the safety or efficacy of a product. Considerations in the use of the medical literature are described in the FDA guidance for industry Providing Clinical Evidence of Effectiveness for Human Drug and Biological Products. In applying these fundamental principles to imaging trials, we recommend that sponsors consider whether the methods section in a relevant literature article describes a prospective protocol in sufficient detail to assess the strengths and weaknesses of the protocol design features discussed in this medical imaging guidance. For example, the design features that are critical include selection of the patient population, clinical setting, image handling, image reading plan for the test product and the standard of truth, use of an accepted standard of truth, the statistical plan, and use of appropriate steps to eliminate bias. As literature studies are often completed for purposes other than drug approval, the relevance of the selected endpoints to the proposed indication should be justified. We recommend that a critical review of the literature present the method used for the literature search, the criteria used to review the data, and the criteria used to determine the applicability of the results. Although we recommend that each article be reviewed and summarized, we also recommend that the key articles be discussed more extensively. Typically, articles in the imaging literature provide limited data on safety, so that additional safety studies may be called for. Other information that can be supplied either fully or partially by the literature include:
In the FDA guidance for industry, Providing Clinical Evidence of Effectiveness for Human Drug and Biological Products, the Agency discusses the use of the medical literature by sponsors. We recommend:
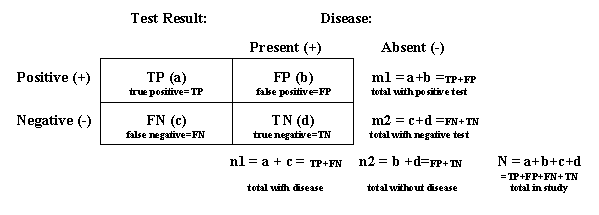
Note: Subjects in trials of medical imaging agents are often classified into one of four groups depending on (1) whether disease is present (often determined with a truth standard or gold standard) and (2) the results of the diagnostic test of interest (positive or negative). The following table identifies the variables that are used to estimate the parameters defined below.
Accuracy: (1) In common usage, accuracy is the quality of being true or correct. (2) As a measure of diagnostic performance, accuracy is a measure of how faithfully the information obtained using a medical imaging agent reflects reality or truth as measured by a truth standard or gold standard. Accuracy is the proportion of cases, considering both positive and negative test results, for which the test results are correct (i.e., concordant with the truth standard or gold standard.) Accuracy = (a+d)/N = (TP+TN)/(TP+FP+FN+TN). Comparator: An established test against which a proposed test is compared to evaluate the effectiveness of the proposed test. A comparator usually means an agent or modality approved for a similar indication. (See also definition for reference product.) Negative predictive value: The probability that a subject does not have the disease when the test result is negative. Synonyms include predictive value negative. Negative predictive value = d/m2 = TN/(TN+FN). By application of Bayes' Rule, the negative predictive value also can be defined as a function of the pretest probability of disease (p), sensitivity, and specificity: negative predictive value = [(1-p) . specificity]/[(1-p) . specificity + p . (1- sensitivity)]. Positive predictive value: The probability that a subject has the disease when the test result is positive. Synonyms include predictive value positive. Positive predictive value = a/m1 = TP/(TP+FP). By application of Bayes' Rule, the positive predictive value also can be defined as a function of pretest probability of disease (p), sensitivity, and specificity: positive predictive value = [(p . sensitivity)/[p . sensitivity + (1-p) . (1- specificity)]. Precision: A measure of the reproducibility of a test, including reproducibility within and across doses, rates of administration, routes of administration, timings of imaging after product administration, instruments, instrument operators, patients, and image interpreters, and possibly other variables. Precision is usually expressed in terms of variability, using such measures as confidence intervals and/or standard deviations. Precise tests have relatively narrow confidence intervals (or relatively small standard deviations). Reference product: An FDA-approved drug product having an indication similar to that of an investigational drug or biological product to which it is being compared for the purpose of evaluating the effectiveness of the investigational drug or biological product. Sensitivity: The probability that a test result is positive when the subject has the disease. Synonyms include true positive rate. Sensitivity = a/n1 = TP/(TP+FN). Specificity: The probability that a test result is negative when the subject does not have the disease. Synonyms include true negative rate. Specificity = d/n2 = TN/(TN+FP). Truth standard (gold standard): An independent method of measuring the same variable being measured by the investigational drug or biological product that is known or believed to give the true value of a measurement.
1 This guidance has been prepared by the Division of Medical Imaging and Radiopharmaceutical Drug Products and the Office of Therapeutics Research and Review in the Center for Drug Evaluation and Research (CDER) at the Food and Drug Administration. 2 In response to the requirements of the Food and Drug Administration Modernization Act of 1997, FDA amended the drug and biologics regulations (21 CFR 315 and 601) by adding provisions for the evaluation and approval of in vivo radiopharmaceuticals used in the diagnosis or monitoring of diseases (64 FR 26657, May 17, 1999). This guidance elaborates on the provisions contained in that regulation. 3 The guidance is not intended to apply to the development of research drugs that do not provide direct patient benefit with respect to diagnosis, therapy, prevention, or prognosis, or other clinically useful information. These include radioactive drugs for research that are used in accordance with 21 CFR 361.1. Section 361.1(a) states that radioactive drugs (defined in 21 CFR 310.3(n)) are generally recognized as safe and effective when administered under specified conditions to human research subjects in the course of a project intended to obtain basic information about the metabolism of a radioactively labeled drug or about human physiology, pathophysiology, or biochemistry. However, if a radioactive drug is used for immediate therapeutic, diagnostic, or similar purposes or to determine the safety and effectiveness of the drug in humans, or if the radioactive drug has a pharmacological effect in the human body, an IND is required. FDA is developing a guidance on determining when research with radioactive drugs may be conducted under § 361.1. The Agency recognizes the potential of imaging agents as research tools for aiding the development of therapeutic drugs, and some of the principles in the guidance may be applicable to such research. Sponsors of such imaging research agents are urged to contact the Division of Medical Imaging and Radiopharmaceutical Drug Products for advice on development of the imaging research agent. 4 21 CFR 315.2 and 601.31. 5 In this guidance, the terms ligand and carrier refer to the entire nonradionuclidic portion of the diagnostic radiopharmaceutical. 6 For approval of a biological license application, the safety of the proposed product must be demonstrated under section 351(a) of the Public Health Service Act (42 U.S.C. 262(a)). 7 See section IV.C for a definition of defined clinical setting. 8 Studying patients with known disease provides information useful in developing a hypothesis for testing in subsequent clinical trials. Typically, such clinical settings are not used to establish efficacy in disease or pathology detection. 9 See section IV.C for a definition of defined clinical setting. 10 Note that use of a defined clinical setting in studies of medical imaging agents also tends to anchor both the pretest probability and the spectrum (e.g., severity or stage) of the disease or condition under study. Thus, when evaluated in a defined clinical setting, diagnostic performance measures that vary with the pretest probability of the disease or condition (e.g., positive and negative predictive values, accuracy), or that can vary with the spectrum of the disease or condition (e.g., sensitivity, specificity, positive and negative predictive values, accuracy) tend to take on values that are relatively constant for that defined clinical setting. See section III B.
|