|
 |
 |
|
 | |||||
| |||||||||
|
|
|
|
|
Guidance for Industry [PDF version of this document]
DRAFT GUIDANCE This guidance document is being distributed for comment purposes only.
For questions regarding this draft document contact Jon E. Clark, 301-594-5613 or Mike Gavini, 301-827-9053. U.S. Department of Health and Human Services Additional copies are available from: (Tel) 301-827-4573 http://www.fda.gov/cder/guidance/index.htm U.S. Department of Health and Human Services
TABLE OF CONTENTS I. INTRODUCTION II. BACKGROUND III. SCOPE IV. CORRELATION OF IN-PROCESS STRATIFIED SAMPLING WITH POWDER MIX AND FINISHED PRODUCT A. Assessment of Powder Mix Uniformity B. Correlation of Powder Mix Uniformity with Stratified In-Process Dosage Unit Data C. Correlation of Stratified In-Process Samples with the Finished Product V. EXHIBIT/VALIDATATION BATCH POWDER MIX HOMOGENEITY VI. VERIFICATION OF MANUFACTURING CRITERIA A. In-Process Dosage Unit Sampling and Analysis B. Criteria to Meet the Readily Pass Classification VII. ROUTINE MANUFACTURING BATCH TESTING METHODS A. Standard Criteria Method (SCM) 1. Stage 1 Test 2. Stage 2 Test B. Marginal Criteria Method (MCM) C. Switching to Standard Test Method from Marginal Test Method VIII. REPORTING THE USE OF STRATIFIED SAMPLING ATTACHMENT 1: VERIFICATION OF MANUFACTURING CRITERIA ATTACHMENT 2: ROUTINE MANUFACTURING BATCH TESTING
I. INTRODUCTION This guidance is intended to assist manufacturers of human drug products in meeting the requirements of 21 CFR 211.110 for demonstrating the adequacy of mixing to ensure uniformity of in-process powder blends and finished dosage units. This guidance describes the procedures for assessing powder mix adequacy, correlating in-process dosage unit test results with powder mix test results, and establishing the initial criteria for control procedures used in routine manufacturing. FDA's guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidances describe the Agency's current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidances means that something is suggested or recommended, but not required. II. BACKGROUND This guidance is the result of an Agency effort to achieve a science-based policy and regulatory enforcement. Experts from industry, academia, and the FDA developed the principles underlying this guidance after extensive public discussion. A brief history of the evolution of this guidance is provided in the following paragraphs. In response to industry concerns regarding regulations for demonstrating the adequacy of in-process powder mixing, the FDA published a draft guidance for industry on blend uniformity analysis in August 1999.2 Comments submitted to the docket resulted in the formation of the Blend Uniformity Working Group (BUWG) by the Product Quality Research Institute (PQRI).3 The PQRI BUWG conducted a public meeting, PQRI Workshop on Blend Uniformity, on September 7 and 8, 2000. Using the consensus reached by participants in this workshop, the BUWG developed a draft recommendation, The Use of Stratified Sampling of Blend and Dosage Units to Demonstrate Adequacy of Mix for Powder Blends. The draft recommendation received examination and peer review in multiple scientific and public venues. In addition, the Advisory Committee for Pharmaceutical Science (ACPS) reviewed the draft recommendation and received public comment during scheduled meetings of the committee.4 The draft recommendation was revised to incorporate the results of peer review and public comment and was presented to CDER's Center Director in final form on December 30, 2002. The recommendation was subsequently published in the PDA Journal of Pharmaceutical Science and Technology.5 This draft guidance reflects CDER's effort to incorporate the draft recommendation into regulatory policy. III. SCOPE Stratified sampling is the process of sampling dosage units at predefined intervals and collecting representative samples from specifically targeted locations in the compression/filling operation that have the greatest potential to yield extreme highs and lows in test results. These test results are used to monitor the manufacturing process output that is most responsible for causing finished product variability. The test results can be used to develop a single control procedure to ensure adequate powder mix and uniform content in finished products. The methods described in this guidance are not intended to be the only methods for meeting Agency requirements to demonstrate the adequacy of powder mix. Traditional powder blend sampling and testing, in conjunction with testing for uniformity of content in the finished product, can be used to comply with current good manufacturing practice requirements (CGMPs). Use of at-, in-, or on-line measurement systems can also be appropriate and are described in other guidance documents.6 This guidance provides recommendations on how to: · Conduct powder blend sampling and analyses. · Establish initial criteria for stratified sampling of in-process dosage units7 and evaluation of test results. · Analyze the stratified samples and evaluate data. · Correlate the stratified sample data with the powder blend data. · Assess powder mix uniformity.
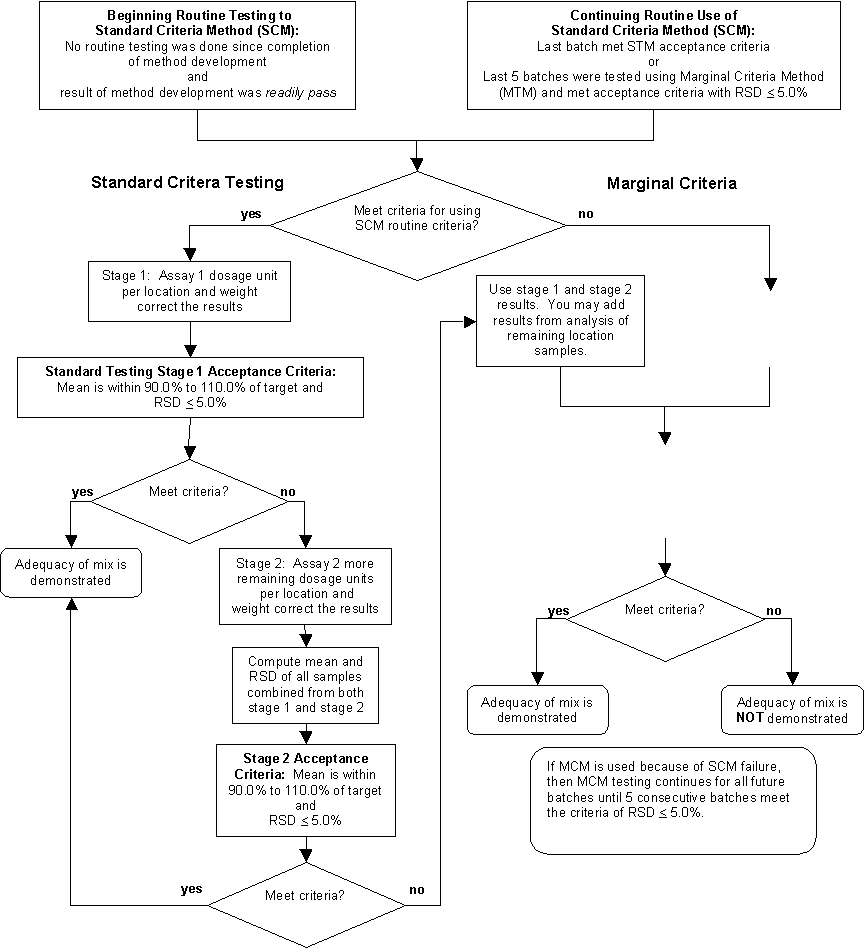
· Test exhibit and validation batches for adequacy of powder mix. · Test and evaluate routine manufacturing batches. · Report the use of stratified sampling in the application. The methods described in this guidance can be used to monitor active ingredient homogeneity of powder blends and to ensure uniform content of the finished product for solid oral drug products. These methods are only one way to satisfy the CGMP and application review requirements for in-process testing to demonstrate adequacy of powder mix and uniform content of the finished product. The method assumes appropriate monitoring of all manufacturing steps as required by the regulations or application commitments. This guidance does not discuss the assessment of the potency and other attributes that can affect the finished dosage units, or the homogeneity of inactive ingredients. Formulations with extremely low dose and/or high potency may call for more rigorous sampling than that described in this guidance to assess the uniformity of powder blends or the uniformity of content of the finished dosage units. When using the methods described in this guidance, certain data or trends may be observed. We recommend that manufacturers scientifically evaluate these types of research data to determine if they affect the quality of a product and, if so, how. The FDA does not intend to inspect research data collected on an existing product for the purpose of evaluating the suitability of proposed methods. Any FDA decision to inspect research data would be based on exceptional situations similar to those outlined in Compliance Policy Guide Sec. 130.300.8 Those data used to support validation or regulatory submissions will be subject to inspection in the usual manner. IV. CORRELATION OF IN-PROCESS STRATIFIED SAMPLING WITH POWDER MIX AND FINISHED PRODUCT If you plan to follow the procedures described in this guidance document, we recommend that you first complete the process development procedures described in this section before using the methods described in sections V, VI, VII. The subsections below describe how to assess the adequacy of powder mix, uniformity of content of the in-process and finished dosage units through correlation and assessment of data from development, validation and manufacturing batches. These procedures can reveal deficiencies in the blending operation that may not have been previously detected. We recommend that manufacturers correct deficiencies in the blending operation before implementing the routine manufacturing control methods described in this guidance. We recommend the assessment of powder mix uniformity using the following procedures: · Conduct blend analysis on batches by extensively sampling the mix in the blender and/or intermediate bulk containers (IBCs). · Identify appropriate blending time and speed ranges, dead spots in blenders, and locations of segregation in IBCs. Determine sampling errors. · Define the effects of sample size (e.g., 1-10X dosage unit range) while developing a technique capable of measuring the true uniformity of the blend. Sample quantities larger than 3X can be used with adequate scientific justification. Appropriate blend sampling techniques and procedures should be developed for each product with consideration to various designs of blend powder sampling and the physical and chemical properties of the blend components. · Design blend-sampling plans and evaluate them using appropriate statistical analyses. · Quantitatively measure any variability that is present among the samples. Attribute the sample variability to either lack of uniformity of the blend or sampling error. Significant within-location variance in the blend data can be an indication of one factor or a combination of factors such as inadequacy of blend mix, sampling error9 or agglomeration.10, 11 Significant between-location variance in the blend data can indicate that the blending operation is inadequate. We recommend the following steps for correlation: · Conduct periodic sampling and testing of the in-process dosage units by sampling them at defined intervals and locations throughout the compression or filling process. Use a minimum of 20 appropriately spaced in-process dosage unit sampling points. There should be at least 7 samples taken from each of these locations for a total minimum of at least 140 samples. · Take 7 samples from each additional location to further assess each significant event,12 such as filling or emptying of hoppers and IBCs, start and end of the compression or filling process and equipment shutdown. This may be accomplished by using process development batches, validation batches, or by using routine manufacturing batches for approved products. · Significant events may also include observations or changes from one batch to another (e.g., batch scale-up and observations of undesirable trends in previous batch data). · Prepare a summary of the data and analysis used to correlate the stratified sampling locations with significant events in the blending process. We recommend you submit this summary with the application as described in section VIII of this guidance. · Compare the powder mix uniformity with the in-process dosage-unit data described above. · Investigate any discrepancies observed between powder mix and dosage-unit data and establish root causes. At least one trouble-shooting guide is available that may be helpful with this task.13 Possible corrections may range from going back to formulation development to improve powder characteristics to process optimization. Sampling problems may also be negated by use of alternate state-of-the-art methods of in situ real-time sampling and analysis. We recommend the following steps: · Conduct testing for uniform content of the finished product using an appropriate procedure or as specified in the Abbreviated New Drug Application (ANDA) or the New Drug Application (NDA) for approved products. · Compare the results of stratified in-process dosage unit analysis with uniform content of the finished dosage units from the previous step. This analysis should be done without weight correction.14 · Prepare a summary of the data and analysis used to conclude that the stratified in-process sampling provides assurance of uniform content of the finished product. We recommend you submit this summary with the application as described in section VIII of this guidance. V. EXHIBIT/VALIDATATION BATCH POWDER MIX HOMOGENEITY This section describes sampling and testing the powder mix of exhibit and process validation batches used to support implementing the stratified sampling method described in this guidance. We recommend that during the manufacture of exhibit and process validation batches, you assess the uniformity of the powder blend, the in-process dosage units, and the finished product independently. We recommend you use the following steps to identify sampling locations and acceptance criteria prior to the manufacture of the exhibit and/or validation batches.15 1. Carefully identify at least 10 sampling locations in the blender to represent potential areas of poor blending. For example, in tumbling blenders (such as V-blenders, double cones, or drum mixers), samples should be selected from at least two depths along the axis of the blender. For convective blenders (such as a ribbon blender), a special effort should be made to implement uniform volumetric sampling to include the corners and discharge area (at least 20 locations are recommended to adequately validate convective blenders). 2. Collect at least 3 replicate samples from each location. Samples should meet the following criteria: · Assay one sample per location (number of samples (n) _ 10) (n = 20 for ribbon blender). · RSD (relative standard deviation) of all individual results _ 5.0 percent. · All individual results are within 10.0 percent (absolute) of the mean of the results. If samples do not meet these criteria, we recommend that you investigate the failure according to the flow chart in Attachment 1. We also recommend that you not proceed any further with implementation of the methods described in this guidance until the criteria are met. Sampling errors may occur in some powder blends, sampling devices, and techniques that make it impractical to evaluate adequacy of mix using only the blend data. In such cases, we recommend that you use in-process dosage unit data in conjunction with blend sample data to evaluate blend uniformity. Some powder blends may present unacceptable safety risk when directly sampled. The safety risk, once described, may justify an alternate procedure. In such cases, process knowledge and data from indirect sampling combined with additional in-process dosage unit data may be adequate to demonstrate the adequacy of the powder mix. Data analysis used to justify using these alternate procedures should be described in a summary report that is maintained at the manufacturing facility. As an alternative, you can substitute the procedures described in the PDA Technical Report No. 25, (see reference in footnote 8) to ensure that the blend is uniform and that the method meets or exceeds the criteria described above. VI. VERIFICATION OF MANUFACTURING CRITERIA You should complete the assessment of powder mix uniformity and correlation of stratified in-process dosage unit sampling development procedures before establishing the criteria and controls for routine manufacturing. We also recommend that you assess the normality and determine RSD from the results of stratified in-process dosage unit sampling and testing that were developed. The RSD value should be used to classify the testing results as either readily pass (RSD _ 4.0%), marginally pass (RSD _ 6.0%) or inappropriate for demonstration of batch homogeneity (RSD > 6.0%). The procedures are discussed in the following sections: We recommend the following steps: · Carefully identify locations throughout the compression or filling operation to sample in-process dosage units. The sampling locations should also include significant process events such as hopper changeover, filling or machine shutdown and the beginning and end of the compression or filling operation.16 There should be at least 20 locations with 7 samples each for a minimum total of 140 samples. These include periodic sampling locations and significant event locations. · Sample at least 7 in-process dosage units from each sampling location. · Assay at least 3 of the 7 and weight correct each result. (The number of samples should be specified and justified for a given product and process.) · Conduct an analysis of the dosage unit stratified sampling data to demonstrate that the batch has a normal distribution of active ingredient. Indications of trends, bimodal distributions, or other forms of a distribution other than normal should be investigated. If these occurrences significantly affect your ability to ensure batch homogeneity, they should be corrected. · Prepare a summary of this analysis. Potential investigation results along with a description of batch normality should be included in the summary. Submit this summary with the application as described in section VIII of this guidance. In addition to this analysis of batch normality, we recommend that you classify the test results as readily pass or marginally pass according to the following procedure: For each separate batch, compare the test results to the following criteria: · For all individual results (for each batch n _ 60) the RSD _ 4.0 percent. · Each location mean is within 90.0 percent to110.0 percent of target strength. · All individual results are within the range of 75.0 percent to 125.0 percent of target strength. If your test results meet these criteria, they are classified as readily pass and you can start routine batch testing using the Standard Verification Method (SVM) described in section VII. If your test results fail to meet these criteria, we recommend that you compare the results with the marginally pass criteria described below. C. Criteria to Meet the Marginally Pass Classification If your dosage unit test results fail to meet the criteria for the readily pass classification, you should assay the remaining dosage units (all 7 units per location) and compare the test results to the following criteria: · For all individual results (for one batch n _ 140) the RSD _ 6.0 percent. · Each location mean is within 90.0 percent to 110.0 percent of target strength. · All individual results are within the range of 75.0 percent to 125.0 percent of target strength. If your test results meet these criteria, results can be classified as marginally pass. If your samples do not meet these criteria, we recommend that you investigate the failure, find justified and assignable cause(s), correct the deficiencies, and repeat the powder mix homogeneity assessment, in-process dosage unit sampling correlation, and initial criteria establishment procedures. The disposition of batches that have failed the marginally pass criteria is outside the scope of this guidance. D. Sample Locations for Routine Manufacturing We recommend that you prepare a summary of the data analysis from the powder mix assessment and stratified sample testing. From the data analysis, you should establish the stratified sample locations for routine manufacturing, taking into account significant process events and their effect on in-process dosage unit and finished dosage unit quality attributes. You should identify at least 10 sampling locations during capsule filling or tablet compression to represent the entire routine manufacturing batch. VII. ROUTINE MANUFACTURING BATCH TESTING METHODS We recommend that you evaluate the routine manufacturing batches against the following criteria after completing the procedures described above to assess the adequacy of the powder mix and uniform content in finished dosage form. These routine manufacturing batch-testing methods include the Standard Criteria Method (SCM) and the Marginal Criteria Method (MCM). The SCM consists of two stages, each with the same accept/reject criteria. The second of the two stages recommends using a larger sample size to meet these criteria. The MCM uses accept/reject criteria that are different from the SCM. If the batch data fail to conform to the SCM criteria, we recommend continued sampling and testing to intensified criteria (MCM). Both verification methods and the procedures for switching from one to the other are detailed below and in the flow chart in Attachment 2. We recommend using the SCM verification method when either of the following conditions is met: · Results of establishing initial criteria are classified as readily pass. · Results of testing to the MCM pass the criteria for switching to the SCM (see section C below). The SCM should meet the same criteria using a different number of sample test results as described below: 1. Stage 1 Test To perform the stage 1 test, we recommend that you (1) collect at least 3 dosage units from each sampling location, (2) assay 1 dosage unit from each location, (3) weight correct the results, and (4) compare the results with the following criteria:
· RSD of all individual results (n _ 10) _ 5.0 percent. · Mean of all results is 90.0 percent to 110.0 percent of target assay. If the results pass these criteria and the adequacy of mix and uniformity of dosage unit content for the batch are adequate, you can use the SCM for the next batch. If test results fail stage 1 criteria, you should conduct extended testing to stage 2 acceptance criteria. 2. Stage 2 Test To perform the stage 2 test, we recommend that you assay the remaining two dosage units (from stage 1) for each sampling location and compute the mean and RSD of data combined from both stage 1 and stage 2. Compare the results with the following criteria: · For all individual results (n _ 30) the RSD _ 5.0 percent. · Mean of all results is 90.0 percent to 110.0 percent of target assay. If your results pass these criteria, the adequacy of mix and uniformity of content for the batch are adequate and you can use stage 1 of SCM for the next batch. If test results fail the criteria, use the MCM described in the next section. After powder mix assessment, in-process dosage unit stratified sampling correlation and initial criteria establishment, we recommend that you use the MCM when either of the following conditions is met: · Results of initial criteria establishment qualified as marginally pass. · Results of initial criteria establishment qualified as readily pass or a batch was tested according to SCM and the test results failed both stage 1 and stage 2 criteria. Then, we recommend you use the weight-corrected results from the stage 2 SCM analysis and compare this with the MVM criteria: · For all individual results (n _ 30) the RSD _ 6.0 percent. · Mean of all results is 90.0 percent to 110.0 percent of target assay. We recommend that all results from analysis of any remaining location samples be computed with the stage 2 SCM data. No test results should be removed from the analysis. If the test results pass these criteria, the adequacy of mix and uniformity of content for the batch are adequate. We recommend that you continue to test routine manufacturing batches with MCM criteria. If the test results fail the criteria, you should no longer use the verification testing methods to ensure adequacy of mixing or uniformity of content until you investigate the failure (per 21 CFR 211.192) to establish justified assignable cause(s), take necessary corrective actions and repeat the powder mix assessment, stratified sample correlation, and initial criteria establishment procedures. It is appropriate to switch to the SCM when the following criterion is met: · Five consecutive batches pass the MCM criteria and result in RSD _ 5.0 percent VIII. REPORTING THE USE OF STRATIFIED SAMPLING This section refers to the scientific data analysis and other information that should be submitted to an NDA or ANDA. Information submitted in the application should include summary reports and scientific analyses or statements about the method being used. The raw data collected to support using this method should be maintained at the manufacturing site. We recommend that you provide the following information in the Manufacturing Process and Process Controls section of the application (CTD17 3.2.P.3.3). · Statement that the methods in this guidance are being used to demonstrate the adequacy of powder mix or a description of alternative methods that demonstrate the adequacy of the powder mix · Summary of data analysis from the powder mix assessment and from stratified sample testing · Summary of the in-process dosage unit stratified sampling data analysis demonstrating a normal distribution of active ingredient in the batch · Summary of the powder mix sampling data analysis demonstrating that it met the minimum criteria for validation and establishing initial criteria We recommend that you provide the following information in the Drug Product Specification section of the application (CTD 3.2.P.4.1): · Statement in the product specification stating that the methods in this guidance are being used to demonstrate finished product uniformity of content or a description of alternative methods used to demonstrate finished product uniformity of content We also recommend that you provide the following information in the Pharmaceutical Development Information section of the application (CTD 3.2.P.2.2):
· Summary of data analysis for correlation of in-process dosage unit stratified sampling with finished product uniformity of content · Summary of data analysis for correlation of powder mix uniformity with in-process dosage unit stratified sampling If you plan on changing the existing controls for adequacy of mix and uniformity of content to the methods described in this guidance, the change should be considered a minor change as described in the postapproval changes guidance.18 We recommend you provide a notice of the change in the next annual report along with the information indicated in section A, above. The raw data collected to support changes can be maintained at the manufacturing site. Absolute as used to define the acceptable range (+/- 10%) in which individual blend sample values must fall and which is independent of the value of the mean. For example, if the mean of all blend samples is 95.0%, the absolute range is 85.0% to105.0%, (not 95.0% +/- 9.5%). Exhibit Batches refer to any batch submitted in support of an NDA or ANDA. This includes bioequivalence, test, and commercial production batches of a drug product. In-process dosage unit is a capsule or tablet as it is formed in the manufacturing process before it is coated or packaged. RSD is relative standard deviation; RSD = [(standard deviation)/(mean)] x 100%. Significant event is any operation during solid dosage production process that can affect the integrity of the in-process materials and, hence, their quality attributes. Transferring powder from a blender to a bin or from the bin to a hopper are two examples of significant events in the blending and compression process. Stratified sampling is the process of collecting a representative sample by selecting units deliberately from various identified locations within a lot or batch, or from various phases or periods of a process to obtain a sample dosage unit that specifically targets locations throughout the compression/filling operation that have a higher risk of producing failing results in the finished product uniformity of content. Target assay is the intended strength or intended amount of active ingredient in the dosage unit. Validation batch is a batch manufactured and tested to verify the proposed routine manufacturing process controls are adequate. Weight correct is a mathematical correction to eliminate the effect of potentially variable tablet weight on measurement of mix adequacy. For example, a tablet with a strength of 19.4 mg and weight of 98 mg = 19.4 ÷ 98 = 0.198 mg/mg. Label claim is 20 mg per each 100 mg tablet, so the weight corrected result is 0.198 ÷ 0.20 * 100 = 99% of target blend assay. ATTACHMENT 1: VERIFICATION OF MANUFACTURING CRITERIA
Blend Sample Criteria: RSD<5.0% and all individuals are within +/- 10% of mean (absolute)1 Dosage Units During Filling or compression take 7 dosage unit samples from each of at least 20 locations
Dosage Unit Readily Pass Criteria: RSD of all individuals <4.0%, Each location mean is within 90.0% - 110.0% of target potency, and all individuals are within 75.0% and 125.0% of target potency2
Dosage Unit Marginally Pass Criteria: RSD of all individuals <6.0%, Each location mean is within 90.0% - 110.0% of target potency, and all individuals are within 75.0% and 125.0% of target potency2 Blend is not uniform or post blending practices are causing segregation 1 Examples of "mean +/- 10% (absolute)" are: If the mean strength = 95%, then the interval is 95% +/- 10%; thus, all individuals must fall within 85.0% to 105.0%. If the mean strength = 103.0%, then the interval is 103.0% +/- 10.0%; thus all individuals must fall within 93.0% to 113.0%. 2 When comparing individual dosage units to 75.0% - 125.0% of target strength, use the as is results (not corrected for weight). ATTACHMENT 2: ROUTINE MANUFACTURING BATCH TESTING Before using this chart to demonstrate adequacy of mix and content uniformity during routine manufacture conduct assess the powder mix, stratified sample correlation and establishes initial criteria. Identify at least 10 sampling locations during filling or compression to represent the entire batch. Remove 3 or more dosage units at each sampling location.
1 This guidance has been prepared by the Office of Pharmaceutical Science and the Office of Compliance in the Center for Drug Evaluation and Research (CDER) at the Food and Drug Administration in cooperation with the Product Quality Research Institute (PQRI) (see footnote 3). This guidance document represents the Agency's current thinking on assessment of the uniformity of powder blends and finished dosage units in the absence of new technology development or implementation. 2 The FDA withdrew the guidance for industry ANDAs: Blend Uniformity Analysis on May 17, 2002. 3 PQRI is a collaborative body involving FDA's Center for Drug Evaluation and Research (CDER), industry, and academia. Since its inception in January 1996, the mission of PQRI has been to generate scientific information in support of regulatory policies through research. Additional information about PQRI is available at www.pqri.org. 4 The PQRI BUWG recommendation appeared on the public ACPS agenda on November 28, 2001 (introduction), May 8, 2002 (distribution and comment), and October 22, 2002 (final comment). 5 G Boehm, J Clark, J Dietrick, L Foust, T Garcia, M Gavini, L Gelber, J Geoffry, J Hoblitzell, P Jimenez, G Mergen, F Muzzio, J Planchard, J Prescott, J Timmermens, and N Takiar, "The Use of Stratefied Sampling of Blend and Dosage Units to Demonstrate Adequacy of Mix for Powder Blends, PDA J. Pharm. Sci Technol,. 57:59-74, 2003. 6 In August 2003, the Agency issued the draft guidance for industry PAT - A Framework for Innovative Pharmaceutical Manufacturing and Quality Assurance. Once finalized, it will represent the Agency's perspective on this issue. 7 The in-process dosage unit is a capsule or tablet as it is formed in the manufacturing process before it is coated or packaged. 8 FDA/ORA Compliance Policy Guide, Sec. 130.300, FDA Access to Results of Quality Assurance Program Audits and Inspections (CPG7151.02) 9 If blend sampling error is detected, more sophisticated, statistical analyses should be applied to assess the situation, such as the use of methods described in J Berman, DE Elinski, CR Gonzales, JD Hofer, PJ Jimenez, JA Planchard, RJ Tlachac, PF Vogel, "Blend Uniformity Analysis: Validation and In-Process Testing." Technical Report No. 25, PDA J Pharm. Sci. Technol. 51(Suppl 3i-iii), S1-99, 1997. 10 OS Sudah, PE Arratia, D. Coffin-Beach, FJ Muzzio, "Mixing of Cohesive Pharmaceutical Formulations in Tote (Bin)-Blenders," Drug Dev. Ind. Pharm, 28(8): 905-918, 2002. 11 V Swaminathan, DO Kildsig, "Polydisperse powder mixtures: effect of particle size and shape on mixture stability," Drug Dev. Ind. Pharm., 28(1):41-48, 2002. 12 A significant event is any operation during the solid dosage production process that can affect the integrity of the in-process materials - see section IX Glossary. 13 JK Prescott, TJ Garcia, "A Solid Dosage and Blend Content Uniformity Troubleshooting Diagram," Pharm. Technol., 25 (3):68-88, 2001. 14 Weight correction is a mathematical correction to eliminate the effect of potentially variable tablet weight on measurement of mix adequacy-see Glossary, Section IX. 15 This is described in Section IV of this guidance. 16 The beginning and end samples are taken from dosage units that would normally be included in the batch. 17 M4Q: The CTD - Quality, one in a series of guidances that provide recommendations for applicants preparing the Common Technical Document for the Registration of Pharmaceuticals for Human Use (CTD) for submission to the FDA. 18 FDA's guidance for industry on Changes to an Approved NDA or ANDA.
Date created: November 10, 2003 |