U.S. Food and Drug Administration • Center for Drug Evaluation
and Research |
U.S. Food and Drug Administration • Center for Drug Evaluation
and Research |
 |
Guidance Document |
|
|
[PDF version of this document]
U.S. Department of Health and Human Services
Food and Drug Administration
Center for Drug Evaluation and Research (CDER)
Center for Biologics Evaluation and Research (CBER)
August 2001
ICH
Additional copies are available from:
Office of Training and Communications
Division of Drug Information, HFD-240
Center for Drug Evaluation and Research
Food and Drug Administration
5600 Fishers Lane
Rockville, MD 20857
(Tel) 301-827-4573
(Internet) http://www.fda.gov/cder/guidance/index.htm
or
Office of Communication, Training and
Manufacturers Assistance, HFM-40
Center for Biologics Evaluation and Research
Food and Drug Administration
1401 Rockville Pike, Rockville, MD 20852-1448
Internet: http://www.fda.gov/cber/guidelines.htm
Fax: 1-888-CBERFAX or 301-827-3844
Mail: the Voice Information System at 800-835-4709 or 301-827-1800
U.S. Department of Health and Human Services
Food and Drug Administration
Center for Drug Evaluation and Research (CDER)
Center for Biologics Evaluation and Research (CBER)
August 2001
ICH
TABLE OF CONTENTS
|
|
INTRODUCTION 1 BACKGROUND 2 The CTD 2 Preparing and Organizing the CTD 2 Module 4. Nonclinical Study Reports 4 Organization and Format of the ICH Guidances for Industry 7 Numbering 7 GENERAL PRINCIPLES 8 Guidances Referenced 8 MODULE 2: NONCLINICAL OVERVIEW 8 MODULE 2: NONCLINICAL WRITTEN AND TABULATED SUMMARIES 11 Guidance on Nonclinical Written Summaries 11 Introduction 11 General Presentation Issues 11 Guidance on Nonclinical Tabulated Summaries 13 2.6. CONTENT OF NONCLINICAL WRITTEN AND TABULATED SUMMARIES 13 2.6.1 INTRODUCTION 13 2.6.2 PHARMACOLOGY WRITTEN SUMMARY 14 2.6.2.1 Brief Summary 14 2.6.2.2 Primary Pharmacodynamics 14 2.6.2.3 Secondary Pharmacodynamics 14 2.6.2.4 Safety Pharmacology 14 2.6.2.5 Pharmacodynamic Drug Interactions 14 2.6.2.6 Discussion and Conclusions 15 2.6.2.7 Tables and Figures 15 2.6.3 PHARMACOLOGY TABULATED SUMMARY (SEE APPENDIX B) 15 2.6.4 PHARMACOKINETICS WRITTEN SUMMARY 15 2.6.4.1 Brief Summary 15 2.6.4.2 Methods of Analysis 15 2.6.4.3 Absorption 16 2.6.4.4 Distribution 16 2.6.4.5 Metabolism (interspecies comparison) 16 2.6.4.6 Excretion 16 2.6.4.7 Pharmacokinetic Drug Interactions 16 2.6.4.8 Other Pharmacokinetic Studies 16 2.6.4.9 Discussion and Conclusions 16 2.6.4.10 Tables and Figures 17 2.6.5 PHARMACOKINETICS TABULATED SUMMARY (SEE APPENDIX B) 17 2.6.6 TOXICOLOGY WRITTEN SUMMARY 17 2.6.6.1 Brief Summary 17 2.6.6.2 Single-Dose Toxicity 18 2.6.6.3 Repeat-Dose Toxicity (including supportive toxicokinetic evaluation) 18 2.6.6.4 Genotoxicity 18 2.6.6.5 Carcinogenicity (including supportive toxicokinetics evaluations) 18 2.6.6.6 Reproductive and Developmental Toxicity (including range-finding studies and supportive toxicokinetics evaluations) 18 2.6.6.7 Local Tolerance 19 2.6.6.8 Other Toxicity Studies (if available) 19 2.6.6.9 Discussion and Conclusions 19 2.6.6.10 Tables and Figures 19 2.6.7 TOXICOLOGY TABULATED SUMMARY (SEE APPENDIX B) 19 MODULE 4: NONCLINICAL STUDY REPORTS 19 4.1 TABLE OF CONTENTS 19 4.2 STUDY REPORTS 19 4.3 LITERATURE REFERENCES 20 APPENDIX A: EXAMPLES OF TABLES AND FIGURES FOR WRITTEN SUMMARIES 21 APPENDIX B: THE NONCLINICAL TABULATED SUMMARIES TEMPLATES 29 APPENDIX C: THE NONCLINICAL TABULALTED SUMMARIES - EXAMPLES 76
Guidance for Industry1 M4S: The CTD - Safety
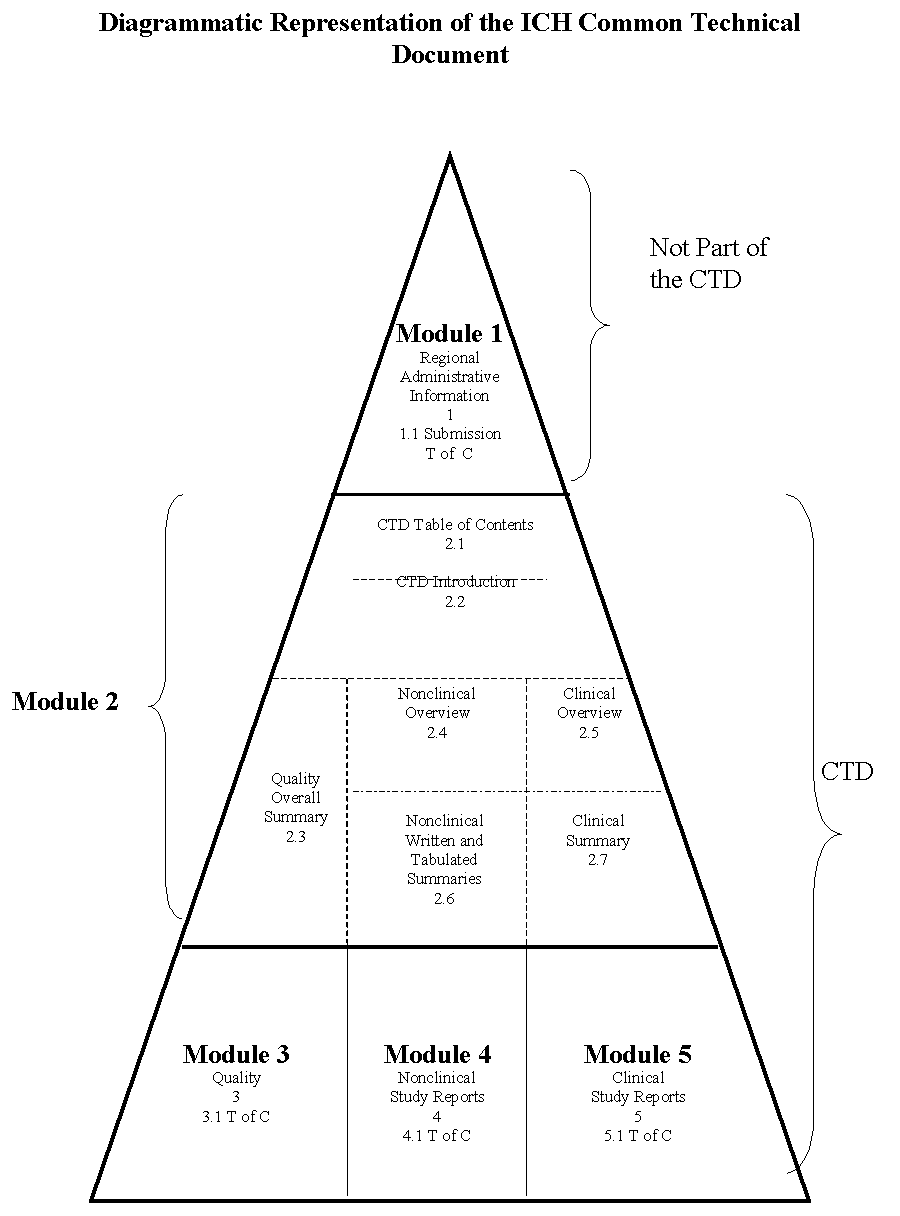
This is one in a series of guidances that provide recommendations for applicants preparing the Common Technical Document for the Registration of Pharmaceuticals for Human Use (CTD) for submission to the U.S. Food and Drug Administration (FDA). This guidance presents the agreed upon common format for the preparation of a well-structured Safety section of the CTD for applications that will be submitted to regulatory authorities. A common format for the technical documentation will significantly reduce the time and resources used to compile applications for registration of human pharmaceuticals and will ease the preparation of electronic submissions. Regulatory reviews and communication with the applicant will be facilitated by a standard document of common elements. In addition, exchange of regulatory information among regulatory authorities will be simplified. For information on the Quality and Efficacy sections of the CTD, see the individual guidances for industry that discuss those parts of the CTD. For general information about the CTD, as well as specific information about Module 1 (regional administrative information), see the guidance for industry, General Considerations for Submitting Marketing Applications According to the ICH/CTD Format.2 The CTD guidances are intended to be used together with other ICH and Agency guidances. Please refer to those guidances for detailed information about the contents of an application. Through the ICH process, considerable harmonization has been achieved among the three regions (Japan, Europe, and the United States) in the technical requirements for the registration of pharmaceuticals for human use. However, until now, there has been no harmonization of the organization of a submission. Each region has its own requirements for the organization of the technical reports in the submission and for the preparation of the summaries and tables. In Japan, the applicants must prepare the GAIYO, which organizes and presents a summary of the technical information. In Europe, expert reports and tabulated summaries are required, and written summaries are recommended. The U.S. FDA has guidance regarding the format and content of the new drug application submission. To avoid generating and compiling different registration dossiers, this guidance describes a format for the Safety section of the CTD that will be acceptable in all three regions. Preparing and Organizing the CTD This guidance primarily addresses the organization of the information to be presented in the Safety section of an application for new pharmaceuticals (including biotechnology-derived products). Guidances also are available that discuss the Quality and Efficacy sections of the CTD. These guidances are not intended to indicate what studies are required. The guidances merely indicate an appropriate format for the data that have been acquired. Applicants should not modify the overall organization of the CTD. However, in the Nonclinical and Clinical Summaries sections of the CTD, applicants can modify individual formats to provide the best possible presentation of the technical information to facilitate the understanding and evaluation of the results. Throughout the CTD, the display of information should be unambiguous and transparent, to facilitate the review of the basic data and to help a reviewer become quickly oriented to the application contents. Text and tables should be prepared using margins that allow the document to be printed on both A4 paper (E.U. and Japan) and 8.5" x 11" paper (U.S.). The left-hand margin should be sufficiently large that information is not obscured through binding. Font sizes for text and tables should be of a style and size that are large enough to be easily legible, even after photocopying. Times New Roman, 12-point font is recommended for narrative text. Acronyms and abbreviations should be defined the first time they are used in each module. References should be cited in accordance with the current edition of the Uniform Requirements for Manuscripts Submitted to Biomedical Journals, International Committee of Medical Journal Editors (ICMJE).3 The CTD should be organized into five modules. Module 1 is region specific. Modules 2, 3, 4, and 5 are intended to be common for all regions. Conformance with the CTD guidances should help ensure that these four modules are provided in a format acceptable to the regulatory authorities (see the figure and overall outline on the following pages). Module 1. Administrative Information and Prescribing Information This module should contain documents specific to each region; for example, application forms or the proposed label for use in the region. The content and format of this module can be specified by the relevant regulatory authorities. For information about this module see the guidance for industry, General Considerations for Submitting Marketing Applications According to the ICH/CTD Format. Module 2. Common Technical Document Summaries Module 2 should begin with a general introduction to the pharmaceutical, including its pharmacologic class, mode of action, and proposed clinical use. In general, the introduction should not exceed one page. Module 2 should contain 7 sections in the following order: · CTD Table of Contents · CTD Introduction · Quality Overall Summary · Nonclinical Overview · Clinical Overview · Nonclinical Written and Tabulated Summaries · Clinical Summary The individual organization of the Module 2 summaries is described in three separate documents: · M4Q: The CTD - Quality · M4S: The CTD - Safety · M4E: The CTD - Efficacy Module 3. Quality Information on Quality should be presented in the structured format described in the guidance, M4Q. Module 4. Nonclinical Study Reports The Nonclinical Study Reports should be presented in the order described in the guidance M4S. Module 5. Clinical Study Reports The human study reports and related information should be presented in the order described in the guidance M4E .
The CTD should be organized according to the following general outline. Module 1: Administrative Information and Prescribing Information 1.1 Table of Contents of the Submission Including Module 1 1.2 Documents Specific to Each Region (for example, application forms, prescribing information) Module 2: Common Technical Document Summaries 2.1 CTD Table of Contents 2.2 CTD Introduction 2.3 Quality Overall Summary 2.4 Nonclinical Overview 2.5 Clinical Overview 2.6 Nonclinical Written and Tabulated Summaries Pharmacology Pharmacokinetics Toxicology 2.7 Clinical Summary Biopharmaceutics and Associated Analytical Methods Clinical Pharmacology Studies Clinical Efficacy Clinical Safety Synopses of Individual Studies Module 3: Quality 3.1 Module 3 Table of Contents 3.2 Body of Data 3.3 Literature References Module 4: Nonclinical Study Reports 4.1 Module 4 Table of Contents 4.2 Study Reports 4.3 Literature References Module 5: Clinical Study Reports 5.1 Module 5 Table of Contents 5.2 Tabular Listing of All Clinical Studies 5.3 Clinical Study Reports 5.4 Literature References Although the CTD is organized by modules, the guidances for industry that provide recommendations for applicants on preparing the CTD have been organized by topic: Quality, Safety, and Efficacy. As a result, guidance discussing Module 2 is divided among three guidances. · Guidance on the Quality section of the CTD (Module 2, Quality Overall Summary (QOS), and Module 3) can be found in the guidance for industry M4Q: The CTD - Quality. · Guidance on the Safety section of the CTD (Module 2, the Nonclinical Overview and the Nonclinical Written and Tabulated Summaries, and Module 4) can be found in the guidance for industry M4S: The CTD - Safety · Guidance on the Efficacy section of the CTD (Module 2, the Clinical Overview and the Clinical Summary, and Module 5) can be found in the guidance for industry M4E: The CTD - Efficacy. In the guidances for industry on the Quality, Safety, and Efficacy sections of the CTD, Arabic numbers have been assigned to designate those specific sections that should be included in the CTD. The Arabic numbers used in the guidances also should be used when assembling the CTD for submission. For specific information on numbering the pages and volumes of the submission, see the guidance for industry, General Considerations for Submitting Marketing Applications According to the ICH/CTD Format. Sections in the guidance documents that are not numbered provide guidance on how to prepare those sections. In this guidance for industry, sections that should be included in Module 2 and Module 4 of the CTD have been numbered using the Arabic numbers 2 and 4, respectively. This guidance provides recommendations for the harmonization of the Safety section of the CTD. It discusses the Nonclinical Overview, Nonclinical Written Summary, and Nonclinical Tabulated Summaries. The primary purpose of the Nonclinical Written and Tabulated Summaries should be to provide a comprehensive, factual synopsis of the nonclinical data. The interpretation of the data, the clinical relevance of the findings, cross-linking with the quality aspects of the pharmaceutical, and the implications of the nonclinical findings for the safe use of the pharmaceutical (i.e., as applicable to labeling) should be addressed in the Nonclinical Overview. The following ICH guidances are referenced in the Safety section: M3 Nonclinical Safety Studies for the Conduct of Human Cllinical Trials for Pharmacaeuticals (November 1997) S1C Dose Selection for Carcinogenicity Studies of Pharmaceuticals (March 1995) S7 Safety Pharmacology Studies for Human Pharmaceuticals (August 2000) MODULE 2: NONCLINICAL OVERVIEW The Nonclinical Overview should provide an integrated, overall analysis of the information in the Common Technical Document. In general, the Nonclinical Overview should not exceed about 30 pages. The Nonclinical Overview should present an integrated and critical assessment of the pharmacologic, pharmacokinetic, and toxicologic evaluation of the pharmaceutical. Where relevant guidances on the conduct of studies exist, these should be taken into consideration, and any deviation from these guidances should be discussed and justified. The nonclinical testing strategy should be discussed and justified. There should be comment on the good laboratory practice (GLP) status of the studies submitted. Any association between nonclinical findings and the quality characteristics of the human pharmaceutical, the results of clinical trials, or effects seen with related products should be indicated, as appropriate. Except for biotechnology-derived products, an assessment of the impurities and degradants present in the drug substance and product should be included, along with what is known of their potential pharmacologic and toxicologic effects. This assessment should form part of the justification for proposed impurity limits in the drug substance and product and be appropriately cross-referenced to the quality documentation. The implications of any differences in the chirality, chemical form, and impurity profile between the compound used in the nonclinical studies and the product to be marketed should be discussed. For biotechnology-derived products, comparability of material used in nonclinical and clinical studies and proposed for marketing should be assessed. If a drug product includes a novel excipient, an assessment of the information regarding the excipient's safety should be provided. Relevant, scientific literature and the properties of related products should be taken into account. If detailed references to published, scientific literature are to be used in place of studies conducted by the applicant, this should be supported by an appropriate justification that reviews the design of the studies and any deviations from available guidances. In addition, the availability of information on the quality of batches of drug substance used in these referenced studies should be discussed. The Nonclinical Overview should contain appropriate reference citations to the Tabulated Summaries in the following format: (Table X.X, Study/Report Number). The Nonclinical Overview should be presented in the following sequence: 2.4. NONCLINICAL OVERVIEW 2.4.1 Overview of the Nonclinical Testing Strategy 2.4.2 Pharmacology 2.4.3 Pharmacokinetics 2.4.4 Toxicology 2.4.5 Integrated Overview and Conclusions 2.4.6 List of Literature Citations Studies conducted to establish the pharmacodynamic effects, the mode of action, and potential side effects should be evaluated, and consideration should be given to the significance of any issues that arise. The assessment of the pharmacokinetic, toxicokinetic, and metabolism data should address the relevance of the analytical methods used, the pharmacokinetic models, and the derived parameters. It might be appropriate to cross-refer to more detailed consideration of certain issues within the pharmacology or toxicology studies (e.g., impact of the disease states, changes in physiology, antiproduct antibodies, cross-species consideration of toxicokinetic data). Inconsistencies in the data should be discussed. Interspecies comparisons of metabolism and systemic exposure comparisons in animals and humans (AUC, Cmax, and other appropriate parameters) should be discussed and the limitations and utility of the nonclinical studies for prediction of potential adverse effects in humans highlighted. The onset, severity, and duration of the toxic effects, their dose dependency and degree of reversibility (or irreversibility), and species- or gender-related differences should be evaluated and important features discussed, particularly with regard to: · Pharmacodynamics · Toxic signs · Causes of death · Pathologic findings · Genotoxic activity - the chemical structure of the compound, its mode of action, and its relationship to known genotoxic compounds · Carcinogenic potential in the context of the chemical structure of the compound, its relationship to known carcinogens, its genotoxic potential, and the exposure data · The carcinogenic risk to humans - if epidemiologic data are available, they should be taken into account · Fertility, embryofetal development, pre- and postnatal toxicity · Studies in juvenile animals · The consequences of use before and during pregnancy, during lactation, and during pediatric development · Local tolerance · Other toxicity studies and/or studies to clarify special problems The evaluation of toxicology studies should be arranged in a logical order so that all relevant data elucidating a certain effect and/or phenomenon are brought together. Extrapolation of the data from animals to humans should be considered in relation to: · Animal species used · Numbers of animals used · Routes of administration employed · Dosages used · Duration of treatment or of the study · Systemic exposures in the toxicology species at no observed adverse effect levels and at toxic doses, in relation to the exposures in humans at the maximum recommended human dose. Tables or figures summarizing this information are recommended. · The effect of the drug substance observed in nonclinical studies in relation to that expected or observed in humans If alternatives to whole animal experiments are employed, their scientific validity should be discussed. The integrated overview and conclusions should clearly define the characteristics of the human pharmaceutical, as demonstrated by the nonclinical studies, and arrive at logical, well-argued conclusions supporting the safety of the product for the intended clinical use. Taking the pharmacology, pharmacokinetics, and toxicology results into account, the implications of the nonclinical findings for the safe human use of the pharmaceutical should be discussed (i.e., as applicable to labeling). MODULE 2: NONCLINICAL WRITTEN AND TABULATED SUMMARIES This guidance is intended to assist authors in the preparation of nonclinical pharmacology, pharmacokinetics, and toxicology written summaries in an appropriate format. This guidance is not intended to indicate what studies are required. It merely indicates an appropriate format for the nonclinical data that have been acquired. The sequence and content of the Nonclinical Written Summary sections are described below. It should be emphasized that no guidance can cover all eventualities, and common sense and a clear focus on the needs of the regulatory authority assessor are the best guides to constructing a document. Therefore, applicants can modify the format, if needed, to provide the best possible presentation of the information and to facilitate the understanding and evaluation of the results. Whenever appropriate, age- and gender-related effects should be discussed. Relevant findings with stereoisomers and/or metabolites should be included, as appropriate. Consistent use of units throughout the Nonclinical Written Summaries will facilitate their review. A table for converting units might also be useful. In the Discussion and Conclusion sections, information should be integrated across studies and across species, and exposure in the test animals should be related to exposure in humans given the maximum intended doses. When available, in vitro studies should precede in vivo studies. Where multiple studies of the same type are summarized within the Pharmacokinetics and Toxicology sections, studies should be ordered by species, by route, and then by duration (shortest duration first). Species should be ordered as follows: · Mouse · Rat · Hamster · Other rodent · Rabbit · Dog · Nonhuman primate · Other nonrodent mammal · Nonmammals Routes of administration should be ordered as follows: · The intended route for human use · Oral · Intravenous · Intramuscular · Intraperitoneal · Subcutaneous · Inhalation · Topical · Other Although the Nonclinical Written Summaries are envisaged to be composed mainly of text, some information contained within them might be more effectively and/or concisely communicated through the use of appropriate tables or figures. Examples of formats that might be included in the Written Summaries are shown in Appendix A. To allow authors flexibility in defining the optimal structure for the written summaries, tables and figures should preferably be included within the text. Alternatively, they could be grouped together at the end of each of the Nonclinical Written Summaries. Throughout the text, reference citations to the Tabulated Summaries should be included in the following format: (Table X.X, Study/Report Number). Although there is no formal limit to the length of the Nonclinical Written Summaries, it is recommended that the total length of the three Nonclinical Written Summaries in general not exceed 100-150 pages. The following order is recommended: · Introduction · Pharmacology written summary · Pharmacology tabulated summary · Pharmacokinetics written summary · Pharmacokinetics tabulated summary · Toxicology written summary · Toxicology tabulated summary It is recommended that summary tables for the nonclinical information in the Common Technical Document be provided in the format outlined in this guidance. Applicants can modify the format, if warranted, to provide the best possible presentation of the information and to facilitate the understanding and evaluation of the results. This guidance is not intended to indicate what studies are requested, but solely to advise how to tabulate study results if a study is performed. Applicants can add some items to or delete some items from the cited format, where appropriate. One tabular format can contain results from several studies. Alternatively, it may be appropriate to cite the data resulting from one study in several tabular formats. The recommended formats for the tables in the Nonclinical Tabulated Summaries are provided in Appendices B and C, which follow. Appendix B contains templates for use in preparation of the tables. The templates are annotated (in italics) to provide guidance on their preparation. (The italicized information should be deleted when the tables are prepared.) Appendix C provides examples of the summary tables. The purpose of the examples is to provide additional guidance on the suggested content and format of the Tabulated Summaries. However, it is the responsibility of the applicant to decide on the best possible presentation of the data for each product. Authors should keep in mind that, in some regions, a review of the Tabulated Summaries (in conjunction with the Written Summaries) represents the primary review of the nonclinical information. Presentation of the data in the formats provided as templates and examples should ensure that a sufficient level of detail is available to the reviewer and should provide concise overviews of related information. When a juvenile animal study has been conducted, it should be tabulated using the template appropriate for the type of study. The order of presentation given for the Nonclinical Written Summaries should be followed for the preparation of the tables for the Nonclinical Tabulated Summaries. 2.6. CONTENT OF NONCLINICAL WRITTEN AND TABULATED SUMMARIES The aim of this section should be to introduce the reviewer to the pharmaceutical and to its proposed clinical use. The following key elements should be covered: · Brief information concerning the pharmaceutical's structure (preferably, a structure diagram should be provided) and pharmacologic properties · Information concerning the pharmaceutical's proposed clinical indication, dose, and duration of use 2.6.2 PHARMACOLOGY WRITTEN SUMMARY Within the Pharmacology Written Summary, the data should be presented in the following sequence: · Brief summary · Primary pharmacodynamics · Secondary pharmacodynamics · Safety pharmacology · Pharmacodynamic drug interactions · Discussion and conclusions · Tables and figures (either here or included in text) The principal findings from the pharmacology studies should be briefly summarized in approximately two to three pages. This section should begin with a brief description of the content of the pharmacologic data package, pointing out any notable aspects such as the inclusion and/or exclusion of particular data (e.g., lack of an animal model). Studies on primary pharmacodynamicsa should be summarized and evaluated. Where possible, it would be helpful to relate the pharmacology of the drug to available data (e.g., selectivity, safety, potency) on other drugs in the class. Studies on secondary pharmacodynamicsa should be summarized by organ system, where appropriate, and evaluated in this section. Safety pharmacology studiesa should be summarized and evaluated in this section. In some cases, secondary pharmacodynamic studies can contribute to the safety evaluation when they predict or assess potential adverse effects in humans. In such cases, these secondary pharmacodynamic studies should be considered, along with safety pharmacology studies. If they have been performed, pharmacodynamic drug interaction studies should be briefly summarized in this section. This section provides an opportunity to discuss the pharmacologic evaluation and to consider the significance of any issues that arise. Text tables and figures can be included at appropriate points throughout the summary within the text. Alternatively, tables and figures can be included at the end of the summary. 2.6.3 PHARMACOLOGY TABULATED SUMMARY (SEE APPENDIX B) 2.6.4 PHARMACOKINETICS WRITTEN SUMMARY The sequence of the Pharmacokinetics Written Summary should be as follows: · Brief summary · Methods of analysis · Absorption · Distribution · Metabolism · Excretion · Pharmacokinetic drug interactions · Other pharmacokinetic studies · Discussion and conclusions · Tables and figures (either here or included in text) The principal findings from the pharmacokinetics studies should be briefly summarized in approximately two to three pages. This section should begin with a description of the scope of the pharmacokinetic evaluation, emphasizing, for example, whether the species and strains examined were those used in the pharmacology and toxicology evaluations, and whether the formulations used were similar or identical. This section should contain a brief summary of the methods of analysis for biological samples, including the detection and quantification limits of an analytical procedure. If possible, validation data for the analytical method and stability of biological samples should be discussed in this section. The potential impact of different methods of analysis on the interpretation of the results should be discussed in the following relevant sections. The following data should be summarized in this section: · Absorption (extent and rate of absorption, in vivo and in situ studies) · Kinetic parameters, bioequivalence and/or bioavailability (serum/plasma/blood PK studies) The following data should be summarized in this section: · Tissue distribution studies · Protein binding and distribution in blood cells · Placental transfer studies The following data should be summarized in this section: · Chemical structures and quantities of metabolites in biological samples · Possible metabolic pathways · Presystemic metabolism (GI/hepatic first-pass effects) · In vitro metabolism including P450 studies · Enzyme induction and inhibition The following data should be summarized in this section: · Routes and extent of excretion · Excretion in milk If they have been performed, nonclinical pharmacokinetic drug interaction studies (in vitro and/or in vivo) should be briefly summarized in this section. If studies have been performed in nonclinical models of disease (e.g., renally impaired animals), they should be summarized in this section. This section provides an opportunity to discuss the pharmacokinetic evaluation and to consider the significance of any issues that arise. Text tables and figures can be included at appropriate points throughout the summary within the text. Alternatively, there is the option of including tables and figures at the end of the summary. 2.6.5 PHARMACOKINETICS TABULATED SUMMARY (SEE APPENDIX B) 2.6.6 TOXICOLOGY WRITTEN SUMMARY The sequence of the Toxicology Written Summary should be as follows: · Brief summary · Single-dose toxicity · Repeat-dose toxicity · Genotoxicity · Carcinogenicity · Reproductive and developmental toxicity · Studies in juvenile animals · Local tolerance · Other toxicity studies · Discussion and conclusions · Tables and figures (either here or included in text) The principal findings from the toxicology studies should be briefly summarized in a few pages (generally not more than six). In this section, the extent of the toxicologic evaluation can be indicated by the use of a table listing the principal toxicologic studies (results should not be presented in this table), for example: Toxicology Program
* This column should be included only if metabolites are investigated. The scope of the toxicologic evaluation should be described in relation to the proposed clinical use. A comment on the GLP status of the studies should be included. The single-dose data should be very briefly summarized, in order by species and by route. In some instances, it may be helpful to provide the data in the form of a table. Studies should be summarized in order by species, by route, and by duration, giving brief details of the methodology and highlighting important findings (e.g., nature and severity of target organ toxicity, dose (exposure) and/or response relationships, no observed adverse effect levels). Nonpivotal studies can be summarized in less detail (pivotal studies are the definitive GLP studies specified by ICH guidance M3). Studies should be briefly summarized in the following order: · In vitro nonmammalian cell system · In vitro mammalian cell system · In vivo mammalian system (including supportive toxicokinetics evaluation) · Other systems A brief rationale should explain why the studies were chosen and the basis for high-dose selection. Individual studies should be summarized in the following order: · Long-term studies (in order by species), including range-finding studies that cannot appropriately be included under repeat-dose toxicity or pharmacokinetics) · Short- or medium-term studies (including range-finding studies that cannot appropriately be included under repeat-dose toxicity or pharmacokinetics) · Other studies 2.6.6.6 Reproductive and Developmental Toxicity (including range-finding studies and supportive toxicokinetics evaluations) Studies should be summarized in the following order, giving brief details of the methodology and highlighting important findings: · Fertility and early embryonic development · Embryofetal development · Prenatal and postnatal development, including maternal function · Studies in which the offspring (juvenile animals) are dosed and/or further evaluated if such studies have been conducted If modified study designs are used, the subheadings should be modified accordingly. If local tolerance studies have been performed, they should be summarized in order by species, by route, and by duration, giving brief details of the methodology and highlighting important findings. If other studies have been performed, they should be summarized. When appropriate, the rationale for conducting the studies should be provided. · Antigenicity · Immunotoxicity · Mechanistic studies (if not reported elsewhere) · Dependence · Studies on metabolites · Studies on impurities · Other studies This section should provide an opportunity to discuss the toxicologic evaluation and the significance of any issues that arise. Tables or figures summarizing this information are recommended. Text tables and figures can be included at appropriate points throughout the summary within the text. Alternatively, tables and figures can be included at the end of the summary. 2.6.7 TOXICOLOGY TABULATED SUMMARY (SEE APPENDIX B)
MODULE 4: NONCLINICAL STUDY REPORTS This guidance presents an agreed upon format for the organization of the nonclinical reports in the Common Technical Document for applications that will be submitted to regulatory authorities. This guidance is not intended to indicate what studies are required. It merely indicates an appropriate format for the nonclinical data that have been acquired. The appropriate location for individual animal data is in the study report or as an appendix to the study report. A Table of Contents should be provided that lists all of the Nonclinical Study Reports and gives the location of each study report in the Common Technical Document. The study reports should be presented in the following order: 4.2.1 Pharmacology 4.2.1.1 Primary Pharmacodynamics 4.2.1.2 Secondary Pharmacodynamics 4.2.1.3 Safety Pharmacology 4.2.1.4 Pharmacodynamic Drug Interactions 4.2.2 Pharmacokinetics 4.2.2.1 Analytical Methods and Validation Reports (if separate reports are available) 4.2.2.2 Absorption 4.2.2.3 Distribution 4.2.2.4 Metabolism 4.2.2.5 Excretion 4.2.2.6 Pharmacokinetic Drug Interactions (nonclinical) 4.2.2.7 Other Pharmacokinetic Studies 4.2.3 Toxicology 4.2.3.1 Single-Dose Toxicity (in order by species, by route) 4.2.3.2 Repeat-Dose Toxicity (in order by species, by route, by duration, including supportive toxicokinetics evaluations) 4.2.3.3 Genotoxicity 4.2.3.3.1 In vitro 4.2.3.3.2 In vivo (including supportive toxicokinetics evaluations) 4.2.3.4 Carcinogenicity (including supportive toxicokinetics evaluations) 4.2.3.4.1 Long-term studies (in order by species, including range-finding studies that cannot appropriately be included under repeat-dose toxicity or pharmacokinetics) 4.2.3.4.2 Short- or medium-term studies (including range-finding studies that cannot appropriately be included under repeat-dose toxicity or pharmacokinetics) 4.2.3.4.3 Other studies 4.2.3.5 Reproductive and Developmental Toxicity (including range-finding studies and supportive toxicokinetics evaluations) (If modified study designs are used, the following subheadings should be modified accordingly.) 4.2.3.5.1 Fertility and early embryonic development 4.2.3.5.2 Embryofetal development 4.2.3.5.3 Prenatal and postnatal development, including maternal function 4.2.3.5.4 Studies in which the offspring (juvenile animals) are dosed and/or further evaluated 4.2.3.6. Local Tolerance 4.2.3.7. Other Toxicity Studies (if available) 4.2.3.7.1 Antigenicity 4.2.3.7.2 Immunotoxicity 4.2.3.7.3 Mechanistic studies (if not included elsewhere) 4.2.3.7.4 Dependence 4.2.3.7.5 Metabolites 4.2.3.7.6 Impurities 4.2.3.7.7 Other 1 This guidance was developed within the Expert Working Group (Safety) of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) and has been subject to consultation by the regulatory parties, in accordance with the ICH process. This document was endorsed by the ICH Steering Committee at Step 4 of the ICH process, November 2000. At Step 4 of the process, the final draft is recommended for adoption to the regulatory bodies of the European Union, Japan, and the United States. A draft version of the General Considerations guidance is currently available. Once it has been finalized, it will represent the Agency's thinking on this topic. 3 The first edition of the Uniform Requirements for Manuscripts Submitted to Biomedical Journals was conceived by the Vancouver Group and was published in 1979.
FDA/Center for Drug Evaluation and Research |