Guidance for
Industry
Pharmacogenomic
Data Submissions
This guidance represents the Food
and Drug Administration's (FDA's) current thinking on this
topic. It does not create or confer any rights for or on any
person and does not operate to bind FDA or the public. You can
use an alternative approach if the approach satisfies the
requirements of the applicable statutes and regulations. If you
want to discuss an alternative approach, contact the FDA staff
responsible for implementing this guidance. If you cannot
identify the appropriate FDA staff, call the appropriate number
listed on the title page of this guidance.
I.
INTRODUCTION
This guidance
is intended to facilitate scientific progress in the field of
pharmacogenomics and to facilitate the use of pharmacogenomic data
in drug development. The guidance provides recommendations to
sponsors holding investigational new drug applications (INDs), new
drug applications (NDAs), and biologics license applications (BLAs)
on (1) when to submit pharmacogenomic data to the Agency during
the drug or biological drug product
development and review processes, (2) what format and content to
provide for submissions, and (3) how and when the data will be
used in regulatory decision making. Key information, including
examples of when pharmacogenomic data submissions would be
required and when voluntary genomic data submissions (VGDSs) would
be welcome are provided in a separate companion document (Pharmacogenomic
Data Submissions, Attachment: Examples of Voluntary Submissions
or Submissions Required Under 21 CFR 312, 314, or 601).
For the
purposes of this guidance, the term pharmacogenomics is
defined as the use of a pharmacogenomic or pharmacogenetic test
(see glossary for definitions) in conjunction with drug therapy.
Pharmacogenomics does not include the use of genetic or genomic
techniques for the purposes of biological product characterization
or quality control (e.g., cell bank characterization, bioassays).
The FDA plans to provide guidance on those uses at a future time.
Pharmacogenomics also does not refer to data resulting from
proteomic or metabolomic techniques. This document is not meant
to provide guidance on pharmacoproteomics or multiplexed protein
analyte based technologies. However, the voluntary submission
process described in this guidance may be used to submit such data
if so desired.
FDA's guidance
documents, including this guidance, do not establish legally
enforceable responsibilities. Instead, guidances describe the
Agency's current thinking on a topic and should be viewed only as
recommendations, unless specific regulatory or statutory
requirements are cited. The use of the word should in
Agency guidances means that something is suggested or recommended,
but not required.
The promise of pharmacogenomics
lies in its potential to help identify sources of inter-individual
variability in drug response (both effectiveness and toxicity);
this information will make it possible to individualize therapy
with the intent of maximizing effectiveness and minimizing risk.
However, the field of pharmacogenomics is currently in early
developmental stages, and such promise has not yet been realized.
The Agency has heard that pharmaceutical sponsors have been
reluctant to embark on programs of pharmacogenomic testing during
FDA-regulated phases of drug development because of uncertainties
in how the data will be used by FDA in the drug application review
process. This guidance is intended to help clarify FDA policy in
this area.
Sponsors
submitting or holding INDs, NDAs, or BLAs are subject to FDA
requirements for submitting to the Agency data relevant to drug
safety and effectiveness (including 21 CFR 312.22, 312.23, 312.31,
312.33, 314.50, 314.81, 601.2, and 601.12). Because these
regulations were developed before the advent of widespread animal
or human genetic or gene expression testing, they do not
specifically address when such data must be submitted. The FDA
has received numerous inquiries about what these regulations
require of sponsors who are conducting such testing.
From a public
policy perspective, a number of factors should be considered when
interpreting how these regulations apply to the developing field
of pharmacogenomics. Because the field of pharmacogenomics is
rapidly evolving, in many circumstances, the experimental results
may not be well enough established scientifically to be suitable
for regulatory decision making. For example:
·
Laboratory techniques
and test procedures may not be well validated. In addition, test
systems may vary so that results may not be consistent or
generalizable across different platforms. A move to standardize
assays is underway, and much more information should be available
within the next several years.
·
The scientific
framework for interpreting the physiologic, toxicologic,
pharmacologic, or clinical significance of certain experimental
results may not yet be well understood.
·
The findings from a
specific study often cannot be extrapolated across species or to
different study populations (e.g., various human subpopulations
with different genetic backgrounds).
·
The standards for
transmission, processing, and storage of the large amounts of
highly dimensional data generated from microarray technology have
neither been well defined nor widely tested.
Despite these
concerns, some pharmacogenetic tests — primarily those related to
drug metabolism — have well-accepted mechanistic and clinical
significance and are currently being integrated into drug
development decision making and clinical practice.
It is
important for FDA to have a role in the evaluation of
pharmacogenomic tests, both to ensure that evolving FDA policies
are based on the best science and to provide public confidence in
the field. The FDA developed this guidance to facilitate the use
of pharmacogenomic tests during drug development and encourage
open and public sharing of data and information on pharmacogenomic
test results.
To this end,
the Agency has undertaken a process for obtaining input from the
scientific community and the public. On May 16 and 17, 2002, the
Agency held a workshop, cosponsored by pharmaceutical industry
groups, to identify key issues associated with the application of
pharmacogenetics and pharmacogenomics to drug development.
Subsequently, on April 8, 2003, a public presentation was made to
the FDA Science Board. This presentation contained a proposal for
developing guidance on the submission of information on
pharmacogenomic tests and a potential algorithm for deciding
whether submission of such data is voluntary or required. The
Science Board endorsed moving forward with both of these
proposals. In November 2003, FDA published a draft version of this
guidance and received public comment on the draft guidance. The
Agency also has developed internal policy related to
pharmacogenomics and voluntary submissions.
The policies
and processes outlined in this final guidance are intended to take
the above factors into account and to assist in advancing the
field in a manner that will benefit both drug development programs
and the public health.
The FDA recognizes that its
pharmacogenomic data submission policies must be consistent with
the relevant codified regulatory submission requirements for
investigational and marketing application submitters and holders.
At present, many pharmacogenomic results are not well enough
established scientifically to be appropriate for regulatory
decision making.
This guidance interprets FDA's regulations for investigational and
marketing submissions, with the goal of clarifying FDA's current
thinking about when the regulations require pharmacogenomic data
to be submitted and when the submission of such data would be
welcome on a voluntary basis. In some cases, complete reports of
pharmacogenomic studies suffice, while in others, an abbreviated
report or synopsis should or must be submitted.
Because FDA regulations establish
different requirements for investigational applications,
unapproved marketing applications, and approved marketing
applications, this guidance sets out different submission
algorithms for each of these categories. The guidance also
clarifies how the Agency currently intends to use such data in
regulatory decision making — that is, when the data will be
considered sufficiently reliable to serve as the basis for
regulatory decision making; when it will be considered only
supportive to a decision; and when the data will not be used in
regulatory decision making.
This guidance also makes a
distinction between pharmacogenomic tests that may be considered
either probable or known valid biomarkers, which may be
appropriate for regulatory decision making, and other less
well-developed tests that are either observational or exploratory
biomarkers that, alone, are insufficient for making regulatory
decisions. Although, currently, most pharmacogenomic measurements
are not considered valid biomarkers, certain markers (e.g., for
drug metabolism) are well established biomarkers with clear
clinical significance. Undoubtedly, the distinction between what
tests are appropriate for regulatory decision making and those
that are not will change over time as the science evolves.
Throughout the development of these tests, as appropriate, FDA
will continue to seek public comment as we evaluate whether a
biomarker is a valid biomarker (e.g., via discussions at
Advisory Committee meetings).
For the purposes of this guidance,
a pharmacogenomic test result may be considered a valid
biomarker if (1) it is measured in an analytical test system
with well-established performance characteristics and (2) there is
an established scientific framework or body of evidence that
elucidates the physiologic, pharmacologic, toxicologic, or
clinical significance of the test results. For example, the
consequences for drug metabolism of genetic variation in the human
enzymes CYP2D6 and thiopurine methyltransferase are well
understood in the scientific community and are reflected in
certain approved drug labels. The results of genetic tests that
distinguish allelic variants of these enzymes are considered to be
well established and, therefore, valid biomarkers.
This guidance makes an additional
distinction between known valid biomarkers that have been accepted
in the broad scientific community and probable valid biomarkers
that appear to have predictive value for clinical outcomes, but
may not yet be widely accepted or independently verified by other
investigators or institutions (see Glossary). When a sponsor
generates, or possesses, data sufficient to establish a
significant association between a pharmacogenomic test result and
clinical outcomes, the test result represents a probable valid
biomarker. It would be expected that this biomarker would meet
criteria (1) and (2) above, and its association with a meaningful
outcome would have been demonstrated in more than one experiment.
The algorithms
described below for investigational and marketing application
holders describe when to submit to FDA data on known valid
biomarkers. Data on probable valid biomarkers need not be
submitted to the IND unless they are used by a sponsor to make
decisions regarding specific animal safety studies or clinical
trials (e.g., using biomarker data as inclusion or exclusion
criteria, assessment of treatment-related prognosis, or
stratifying patients by dose) or are a probable valid biomarker in
human safety studies (see section IV.A).
However, we recommend that sponsors or applicants submit reports
on all probable valid biomarkers to new (i.e., unapproved) NDAs or
BLAs according to the algorithm in section IV.B.
Many
pharmacogenomic testing programs implemented by pharmaceutical
sponsors or by scientific organizations are intended to develop
the knowledge base necessary to establish the validity of new
genomic biomarkers. During such a period of scientific
exploration, test results are not useful in making regulatory
judgments pertaining to the safety or effectiveness of a drug and
are not considered known or probable valid biomarkers. However,
scientific development of this sort is highly desirable for
advancing the understanding of relationships between genotype or
gene expression and responses to drugs and, therefore, should be
encouraged and facilitated. For these reasons, although
submission of exploratory pharmacogenomic data is not required
under the regulations, FDA is encouraging voluntary submission
of such data, as described below.
As the field
of pharmacogenomics advances, it is likely (and desirable) that
sponsors will begin to use pharmacogenomic tests to support drug
development and/or to guide therapy. Sponsors may choose to
submit pharmacogenomic data that have not achieved the status of a
valid biomarker to an investigational or marketing application to
support scientific contentions related to dosing and dosing
schedule, safety, or effectiveness. For example, a sponsor may
wish to provide supportive data demonstrating that changes in
drug-induced gene expression differ between species that have
different toxicologic responses to a drug, thus correlating
changes in certain gene expression patterns with a specific
toxicity. Or, a pharmacogenomic test result might also be used to
stratify patients in a clinical trial or to identify patients at
higher risk for an adverse event to correlate test results with
clinical outcome.
When
pharmacogenomic results affect the design of a specific animal
safety trial, or human safety or efficacy trial, the submission
algorithms described below suggest that full information on the
test system must be submitted to the IND (§§ 312.30(b) and
312.31). In contrast, results from earlier feasibility studies
done under the same IND (or outside the IND) to establish the
potential usefulness of the pharmacogenomic test (e.g., from
samples taken during a dose-response study) are not a required
submission, but would be encouraged as a voluntary submission.
However, a plan to perform any invasive test, including
phlebotomy, with the possible intent to conduct pharmacogenomic
testing on a sample, must be noted both in the protocol and the
informed consent document (§§ 312.23(a)(6), 312.30(b), and 50.25).
If a
pharmacogenomic test shows promise for enhancing the dose
selection, safety, or effectiveness of a drug, a sponsor may wish
to fully integrate pharmacogenomic data into the drug development
program. This integration could occur in two ways:
1. The pharmacogenomic data
may be intended to be included in the drug labeling in an
informational manner.
For example, such data might be
used to describe the potential for dose adjustment by drug
metabolism genotype (e.g., CYP2D6*5) or to mention the possibility
of a side effect of greater severity or frequency in individuals
of a certain genotype or gene expression profile. In such cases,
the pharmacogenomic test result would be considered a known valid
biomarker. However, an FDA-approved pharmacogenomic test may not
be available or required to be available, or a commercial
pharmacogenomic test may not be widely available. Given this
level of complexity, at the current time, sponsors should consult
the relevant FDA review division for advice on how to proceed in a
specific case. However, whenever a sponsor intends to include
pharmacogenomic data in the drug label, complete information on
the test and results must be submitted to the Agency as described
under §§ 314.50 and 601.2.
2. The pharmacogenomic data
and resulting test or tests may be intended to be included in the
drug labeling to choose a dose and dose schedule, to identify
patients at risk, or to identify patient responders. Inclusion
of a pharmacogenomic test in the labeling would be contingent upon
its performance characteristics. For example:
·
Patients will be
tested for drug metabolism genotype and dosed according to the
test results.
·
Patients will be
selected as potential responders for an efficacy trial (or
deselected because of a high risk) based on genotype (e.g., of
either the patient or the patient’s tumor) or gene expression
profile.
·
Patients will be
excluded from a clinical trial based on genotype or gene
expression profile (e.g., biomarker for risk of an adverse event).
In all of these cases, FDA
recommends co-development of the drug and the pharmacogenomic
tests, if they are not currently available, and submission of
complete information on the test/drug combination to the Agency.
The FDA plans to issue further guidance on co-development of
pharmacogenomic tests and drugs.
The
Office of In Vitro Diagnostics in CDRH, appropriate review
divisions in CBER, and the Clinical and Clinical Pharmacology
Review divisions in CBER or CDER are willing to meet jointly with
sponsors to discuss both scientific and regulatory issues with
regard to new pharmacogenomic tests. The CDRH has both formal
(IDE) and informal (pre-IDE) processes to evaluate protocols for
pharmacogenomic test development.
At the current time, most
pharmacogenomic data are of an exploratory or research
nature, and FDA regulations do not require that these data be
submitted to an IND, or that complete reports be submitted to an
NDA or BLA. However, voluntary submissions can benefit both the
industry and FDA in a general way by providing a means for
sponsors to ensure that regulatory scientists are familiar with
and prepared to appropriately evaluate future genomic
submissions. The FDA and industry scientists alike would benefit
from an enhanced understanding of relevant scientific issues, such
as the following:
·
The types of genetic
loci or gene expression profiles being explored by the
pharmaceutical industry for pharmacogenomic testing
·
The test systems and
techniques being employed
·
The problems
encountered in applying pharmacogenomic tests to drug development
·
The ability to
transmit, store, and process large amounts of complex
pharmacogenomic data streams with retention of fidelity
·
The scientific
rationale for standardizing naming and characterization of the
genes used on different genomic analysis platforms and for
developing bioinformatics software programs used to evaluate
pharmacogenomic data
·
Facilitate
identification of predictors of safety, effectiveness, or toxicity
A greater understanding of the
issues surrounding the use of pharmacogenomic data may prevent
delays in reviews of future submissions where genomics are an
integral part of specific studies in a drug development program.
Therefore, FDA
is requesting that sponsors conducting such programs consider
providing pharmacogenomic data to the Agency voluntarily,
when such data are not otherwise required under the regulations.
To facilitate VGDSs, FDA has established a cross-center
Interdisciplinary Pharmacogenomic Review Group (IPRG) to review
VGDSs, to work on policy development, and, upon request, to advise
review divisions on interpretation and evaluation of
pharmacogenomic data.
For sponsors,
voluntary submission of genomic data offers a number of specific
potential benefits:
·
Meet informally
with FDA and receive peer review assessments of scientific
data from pharmacogenomic experts at the Agency
·
Obtain insight into
the evolving regulatory decision making process as it relates to
genetic and genomic information
·
Familiarize FDA
scientists with novel pharmacogenomic experiments, data analysis,
and interpretation approaches at an early stage
·
Conserve time and
resources by obtaining feedback from FDA on a VGDS that might
highlight unaddressed issues that could prove time consuming or
costly later during product development
·
Identify new
opportunities for drug development (e.g., feedback from FDA might
help reach new strategic decisions). For example, a shelved
product may be continued when new tools such as genotyping assays
become available to demonstrate effectiveness in a subpopulation.
·
Make a contribution
to the VGDS data repository to facilitate advancement of
pharmacogenomics and development of rational, data-based policies
and guidances
The FDA's regulations establish
different requirements for INDs, new (i.e., unapproved) NDAs and
BLAs, and approved NDAs and BLAs. For this reason, there are
different submission algorithms for the submission of
pharmacogenomic data.
Section 312.23 describes
information submission requirements for an IND, including data
generated or available during the IND phase. Section 312.23(a)(8)
contains the requirements for pharmacology and toxicology
information: “Adequate information about pharmacologic and
toxicological studies of the drug involving laboratory animals or
in vitro, on the basis of which the sponsor has concluded
that it is reasonably safe to conduct the proposed clinical
investigations” (emphasis added). The in vitro and animal studies
needed to establish a basis for proceeding with human trials of
various types are well established internationally. Therefore,
pharmacogenomic data relevant to, or derived from, animal or in
vitro studies must ordinarily be submitted according to §
312.23(a)(8) when the sponsor wishes to use these data to make a
scientific case, or when the pharmacogenomic test is a known valid
biomarker.
Section
312.23(a)(9) sets forth the requirements for submitting previous
human experience with an investigational drug. The application
must include a summary of trials or human experience relevant to
an evaluation of the safety or effectiveness of a drug.
Therefore, sponsors must submit human data of known relevance
(e.g., known valid pharmacogenomic biomarkers). In addition,
sponsors or applicants must submit "any other information that
would aid evaluation of the proposed clinical investigations with
respect to their safety or their design and potential as
controlled clinical trials to support the marketing of the drug"
(§ 312.23(a)(10)(iv)). Sponsors may possess human data that
suggest that a particular biomarker is a probable valid biomarker
for evaluating the safety of the drug being evaluated. In these
cases, information on the biomarker must be submitted to the IND
because it could potentially aid in evaluation of the safety of
the investigations per the regulations.
In addition,
section 312.23(a)(11) states that a sponsor must submit "if
requested by FDA, any other relevant information needed for review
of the application." Therefore, during the IND review, FDA may
request pharmacogenomic information the Agency considers relevant
(e.g., information related to the mechanism of action of the
drug).
Sponsors
holding INDs who generate or possess pharmacogenomic data related
to an investigational drug can comply with FDA requirements using
the following algorithm:
Pharmacogenomic data must be submitted to the IND under § 312.23
if ANY of the following apply:
1. The test results are used for
making decisions pertaining to a specific clinical trial, or in an
animal trial used to support safety (e.g., the results will affect
dose and dose schedule selection, entry criteria into a clinical
trial safety monitoring, or subject stratification).
2. A sponsor is using the test
results to support scientific arguments pertaining to, for
example, the pharmacologic mechanism of action, the selection of
drug dosing and dosing schedule, or the safety and effectiveness
of a drug.
3. Test results constitute a
known valid biomarker for physiologic, pathophysiologic,
pharmacologic, toxicologic, or clinical states or outcomes in
humans, or the test is a known valid biomarker for a safety
outcome in animal studies. If the information on the biomarker
(example, human CYP2D6 status) is not being used for
purposes 1 or 2 above, the information can be submitted to the IND
as an abbreviated report.
Submission to an
IND is NOT required, but voluntary
submission is encouraged (i.e., information does not meet the
criteria of § 312.23) if
4. Information is from
exploratory studies or is research data, such as from general gene
expression analyses in cells/animals/humans, or single-nucleotide
polymorphism (SNP) analysis of trial participants.
5. Information consists of
results from test systems where the validity of the biomarker is
not established.
Although
submission of such data in cases 4 and 5 is not required under the
regulations, FDA would welcome voluntary submission of the data in
a VGDS. See Appendix A for additional guidance on assessing
whether to submit pharmacogenomic data to an IND.
Note:
Regardless of requirements for submission, the fact that samples
will be collected for potential analysis must be noted in any
clinical protocol (§ 312.23(a)(6)) and informed consent documents
(§ 50.25).
Data from a
VGDS submission concerning a product under an IND will not be used
for regulatory decision making. However, after the sponsor
submits a VGDS, if additional information becomes available that
triggers the requirements for submission under §§ 312, 314, or
601, the sponsor must submit the data to the relevant application
and should follow the appropriate algorithm.
Section 314.50 outlines the NDA
submission requirements; section 601.2 generally outlines BLA
submission requirements. As the introduction to § 314.50 states,
“the [NDA] application is required to contain reports of all
investigations of the drug product sponsored by the applicant, and
all other information about the drug product pertinent to an
evaluation of the application that is received or otherwise
obtained by the applicant from any source.” Therefore, to comply
with these regulations, sponsors must provide reports of certain
pharmacogenomic investigations in their NDAs, and to permit a
thorough analysis of a biologics application, a sponsor must
submit such a report in its BLA. However, the extent and format
of such reports will depend on the relevance and application of
the information.
Subsequent paragraphs of § 314.50
outline the submission requirements in specific disciplines.
Nonclinical pharmacology and toxicology submission requirements
are described in § 314.50(d)(2); human pharmacokinetics and
bioavailability requirements in § 314.50(d)(3); and clinical data
requirements in § 314.50(d)(5).
Section 601.2
generally outlines the BLA submission requirements. Section 601.2
states that the BLA manufacturer shall submit data derived from
nonclinical laboratory and clinical studies that demonstrate that
the manufactured product meets prescribed requirements of safety.
Like NDA sponsors, BLA sponsors must provide reports of certain
pharmacogenomic investigations in their BLAs. However, the extent
and format of such reports will depend on the relevance and
application of the information.
Sponsors who
have generated or possess pharmacogenomic data related to a drug
can comply with the regulations' requirements using the algorithm
below describing what kind of report to submit:
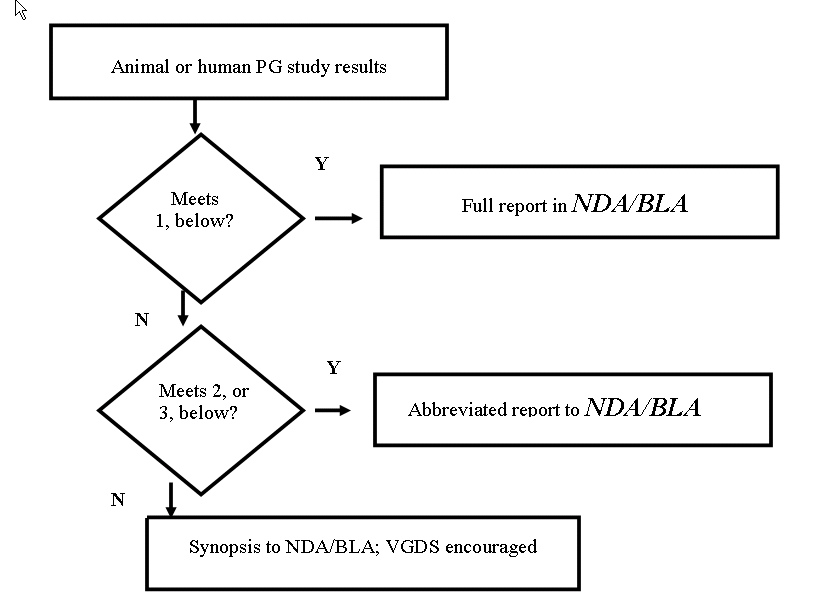
1. Provide
full (complete) reports on pharmacogenomic investigations intended
by the sponsor to be used in the drug label or as part of the
scientific database being used to support approval as complete
submissions (not in the form of an abbreviated report, synopsis,
or VGDS), including information about test procedures and complete
data, in the relevant sections of the NDA or BLA. If the
pharmacogenomic test is already approved by FDA or is the subject
of an application submitted to the Agency, information on the test
itself can be provided by cross reference.
The following examples would
fit this category.
·
Pharmacogenomic test
results from clinical trials used to support scientific arguments
made by the sponsor about selecting drug doses, assessing safety,
selecting patients for treatment, or monitoring the beneficial
responses
·
Pharmacogenomic test
results that the sponsor proposes to describe in the drug labeling
·
Pharmacogenomic tests
that are essential to achieving the dosing, safety, or
effectiveness described in the drug labeling
2. Submit
reports of pharmacogenomic test results that constitute known
valid biomarkers for physiologic, pathophysiologic, pharmacologic,
toxicologic, or clinical states or outcomes in the relevant
species, but that the sponsor is not relying on or mentioning in
the label, to the Agency as an abbreviated report (not in the form
of a synopsis or VGDS). (If a pharmacogenomic test of this type
was conducted as part of a larger overall study, the reporting of
the pharmacogenomic test results can be incorporated into the
larger study report.)
3. Submit
reports of pharmacogenomic tests that represent probable valid
biomarkers for physiologic, pathophysiologic, pharmacologic,
toxicologic, or clinical states or outcomes in the relevant
species to the NDA or BLA as an abbreviated report. (If the
pharmacogenomic testing of this type was conducted as part of a
larger study, the abbreviated report can be appended to the report
of the overall study.)
4. There
is no need to submit detailed reports of general exploratory or
research information, such as broad gene expression screening,
collection of sera or tissue samples, or results of
pharmacogenomic tests that are not known, or probable valid
biomarkers to the NDA or BLA. Because the Agency does not view
such studies as germane in determining the safety or effectiveness
of a product, the submission requirements in §§ 314.50 or 601.2
will be satisfied by the submission of a synopsis of the study.
However, the Agency encourages the voluntary submission of the
data from such a study in a VGDS.
See Appendix B
for additional guidance on how to assess whether to submit
pharmacogenomic data to an unapproved NDA or BLA.
The
requirements for submitting new scientific information to a
previously approved NDA or BLA are outlined in §§ 314.81(b)(2) and
601.12. Results of nonclinical or clinical pharmacogenomic
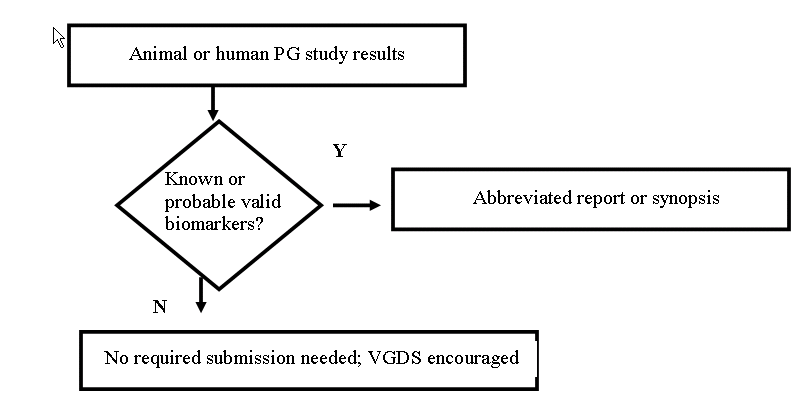
investigations on known or probable valid biomarkers must be
submitted in the annual report as synopses or abbreviated reports
(§ 314.81(b)(2)).
Pharmacogenomic study results of other types do not meet the
submission requirements outlined in the regulations (§
314.81(b)(2)). However, such reports can be voluntarily submitted
to the NDA or BLA as a VGDS.
Pharmacogenomic data collected in pharmacoepidemiologic and
observational studies can be submitted as a VGDS by the applicant
in accordance with the recommendations in this guidance (see
Section VI).
Questions have been raised about
the need for pharmacogenomic studies to comply with the
requirements of 21 CFR part 58, which describes good laboratory
practices (GLPs) for nonclinical laboratory studies that support
INDs and NDAs. Section 58.3(d) (21 CFR 58.3(d)) defines
nonclinical laboratory studies as “in vivo or in vitro
experiments in which test articles are studied prospectively in
test systems under laboratory conditions to determine their
safety. The term does not include studies using human subjects or
clinical studies or field trials in animals. The term does not
include basic exploratory studies carried out to determine whether
a test article has any potential utility….”
The
requirements of part 58 apply to nonclinical studies submitted to
support safety findings, including nonclinical pharmacogenomic
studies intended to support regulatory decision making. If full
compliance with 21 CFR Part 58 cannot be met, a sponsor must
clearly indicate in the study report the areas in which such data
do not comply with Part 58 (§§ 312.23(a)(8)(iii) and
314.50(d)(2)(v)). Any studies eligible to be submitted in an
abbreviated report, synopsis, or VGDS under the algorithms
discussed above do not fall under part 58.
The FDA recognizes that it may not
be feasible to conduct separate, long-term, non-GLP preclinical
studies. For this reason, FDA encourages sampling of tissues from
GLP studies for investigational purposes. Removal of tissue
samples and the reason for removal (e.g., exploratory, mechanistic
study, tissue banking) should be specified in the protocol.
Removal of specimens for investigational purposes from a study
does not invalidate the GLP status of the main toxicology study,
if otherwise acceptable. If the tissue samples are subsequently
analyzed, the results should be reported to the NDA as a
synopsis. The FDA would also be interested in receiving these
data in a VGDS. If findings from these studies are considered by
the sponsor to be relevant to the safety of the compound under
study (e.g., related to a known valid biomarker), the findings
must be reported to the application, as is necessary for any other
relevant nonclinical study findings
312.23(a)(8), 312.32(c)(1)(i)(B),
314.50(d)(2)).
The FDA will also accept
pharmacogenomic data from investigators who may not have an active
IND, NDA, or BLA , but who wish to provide the information
voluntarily to FDA, according to the process described in Section
VI of this guidance.
We recommend
that all VGDSs be prominently marked as VGDS, or
VOLUNTARY SUBMISSIONS, on the cover letter that accompanies
the submission (see Appendix E).
The FDA
invites submission of exploratory pharmacogenomic data on drugs or
candidate drugs whether or not the molecules are currently the
subject of an active IND, NDA, or BLA. Exploratory genomic data
may result from, for example, microarray expression profiling
experiments, genotyping or single-nucleotide polymorphism (SNP)
profiling experiments, or from other studies using evolving
methodologies that are intended to facilitate global analysis of
gene functions, but not specific claims pertaining to drug dosing,
safety assessments, or effectiveness evaluations. Currently,
consensus standards do not exist for presenting and exchanging
genomic data, although such standards are evolving. Therefore,
this guidance does not recommend a specific data format for the
VGDS.
We recommend
that, to achieve the goals of the VGDS process as delineated in
Section III(C), the content of a VGDS, and the level of detail, be
sufficient for the Agency to interpret the information and
independently analyze the data, verify results, and explore
possible genotype-phenotype correlations across studies. We do
not, however, want the submission of a VGDS to be overly
burdensome and time-consuming for sponsors. Therefore, VGDS could
be submitted in a number of forms:
·
As an article
submitted to a peer-reviewed scientific journal with raw or
processed data submitted electronically
·
As an evolving public
standard for specific types of experiments, such as the Minimum
Information About a Microarray Experiment (MIAME) standard for
microarray expression data.
Using an approach similar in content to MIAME one can format a
VGDS containing genotyping or other genomic data derived from
technology platforms other than nucleic acid hybridization arrays.
·
As a full report on a
gene expression microarray experiment, the content could contain
the following analytical, preclinical and/or clinical information,
for example:
– Title page
– Table of contents
– Background and scientific
rationale
– Primary and secondary study goals
– Synopses and summary of findings
– Study design and sample
collection
– Array design and description
- Sample processing and preparation
- Demonstration of quality of RNA
or DNA
- Hybridization procedures and
parameters
- Measures of performance of
hybridization such as spike-in control
- Measurements and quantification
- Normalization controls
- Number of repeats (array
hybridized), number of biological assays performed
– Data Analysis
- Statistical analysis
- Bioinformatics tools and software
used. Source of gene annotation
– Results and conclusions,
including, for example, data visualization (e.g., scatter plots,
principle component analysis (PCA), hierarchical clustering (heat
maps)), correlation between expression profiles and outcomes, and
appropriate information about relevant co-factors
– References
·
Additional Study
Information related to mircroarray studies might include the
following:
–
Confirmation of SNP analysis by sequencing or other assays
-
Confirmation of gene expression by other conventional assays
(e.g., Northern blot, RT-PCR (real time polymerase chain
reaction)). As much as possible, all genes of importance should
be confirmed with secondary assays. However, if the genomic
profile is of importance, it may be appropriate to sample a
selected subpopulation of affected genes
-
Alternative approaches that examine endpoints other than gene
expression changes may also be appropriate under certain
circumstances (e.g., immunohistochemistry or Western blot, if
reagents available).
Using the decision trees (see
Appendices A-C), sponsors should submit genomic data according to
the following recommendations.
·
For required
submissions, complete reports, abbreviated reports, or synopses of
pharmacogenomic studies should be submitted to INDs, NDAs, or BLAs
in the usual manner.
·
For candidate drugs
or stand alone voluntary submissions (submissions not related to
any application), sponsors should submit the package clearly
labeled as VOLUNTARY GENOMIC DATA SUBMISSION (VGDS).
A voluntary submission cover sheet that can be used is
included in Appendix E. For VGDSs related to an existing IND, NDA,
or BLA, please include the reference number on the voluntary
submission cover sheet.
The FDA has received many questions
about the use of pharmacogenomic data in the application review
process. Questions reflect the concern that the Agency will raise
new questions and require additional data based on findings from
exploratory pharmacogenomic studies, that new studies will be
required or suggested based on preliminary human pharmacogenomic
data, that indicated populations will be narrowed or restricted
based on the pharmacogenomic results in subpopulations, or that
new studies in subpopulations will be required after retrospective
analysis suggests differential responses based on pharmacogenomic
subgrouping. There is also concern about the availability of
staff who are experts in interpretation of such data.
The FDA
will not use genomic information submitted through the voluntary
process for regulatory decision making on INDs, BLAs, or NDAs.
VGDSs will be
reviewed by the Interdisciplinary Pharmacogenomic Review Group (IPRG).
The review process is intended to ensure that scientific staff
experienced in the evaluation of genomics studies participate
first-hand in analysis and review of the data. Any data
evaluation will be conducted for scientific and informational
purposes — not for regulatory decision making. If additional
information becomes available after a sponsor submits a VGDS that
triggers the submission requirements under §§ 312, 314, or 601,
the sponsor must resubmit the data to the investigational or
marketing application and should follow the appropriate algorithm
described in this guidance for a required submission. Also, a
review division may consult the IPRG when pharmacogenomic data are
submitted as part of an IND, NDA, or BLA.
The animal and
in vitro toxicology database needed to support human trials at
various stages of the IND process and to support marketing of
short- or long-term use drugs is well established. Any proposals
for the substitution or addition of new animal genomic safety
tests will ordinarily be the product of a public process involving
the international scientific and drug development communities. If
FDA becomes aware that a particular pharmacogenomic test has taken
on great significance based upon cumulative experience (e.g., from
evaluating results across submissions, and/or obtaining input from
Advisory Committees), the Agency will notify sponsors about its
findings.
Currently, as
discussed above, only a few pharmacogenetic tests for certain drug
metabolizing enzymes are considered known valid biomarkers in
humans. Considerable concern has been expressed about how FDA
will evaluate newer types of pharmacogenomic data (e.g., results
that may predict increased risk of adverse events, or point to an
enhanced probability of effectiveness response). The FDA has
considerable experience dealing with these issues in other
contexts. Examples of how pharmacogenomic studies fit into this
experience include the following.
·
Descriptions of drug
metabolizing phenotypes and discussion of their effects on dosing
are common in drug labels. Extrapolation of this information to
pharmacogenetic testing is straightforward.
·
There are many
conditions or co-factors that may increase an individual’s
susceptibility to an adverse event (e.g., co-morbid conditions,
metabolic susceptibilities such as hepatic failure, or concomitant
drug therapies) or the probability of a beneficial response.
The FDA’s usual approach in such
cases has been to request that information be added to the drug
labeling that describes the possible interaction and relevant
co-factors and advises on precautions. If a sponsor discovers a
new pharmacogenomic test that could possibly distinguish patients
at greater risk for a serious adverse event, it is likely that
both the sponsor and the Agency would have great interest in
exploring the correlation in the appropriate populations.
However, if the sponsor also moved forward on developing the drug
in the overall indicated population, FDA would evaluate the safety
database on its merits. If the sponsor decided to develop the
drug solely in populations from which certain patients were
excluded based on pharmacogenomic testing, FDA would recommend
co-development of the pharmacogenomic test (as a diagnostic) and
the drug because FDA would be unable to approve a drug for which
the risk or benefit was predicated on a pharmacogenomic test that
was unavailable.
It is most
likely that, in the near future, pharmacogenomic biomarkers that
predict drug toxicity will be identified and developed on a path
parallel with overall drug development. In other words, a drug
would be developed in a conventional manner with a parallel effort
to identify appropriate predictors of toxicity. If the drug’s
risk-benefit profile were acceptable in the entire target
population, the drug could be approved prior to the completion of
efforts to refine and develop the relevant pharmacogenomic tests.
When and if a test's predictive values were to be established and
the test were to become commercially available (either as an
approved device or as a service), the drug label could be changed
to reflect the data.
·
The FDA has similar
experience with tests used to target populations likely to respond
to therapy.
Several
decades ago, broad indications for use were described in labels.
Over time, as more exact diagnoses were developed, narrower
indications were sought by sponsors, based on the clinical trials
conducted. A similar evolution occurred in the field of anti-HIV
therapies as drug resistance testing became available. We
encourage sponsors to continue to develop pharmacogenomic tests
that are predictive of subpopulations with enhanced response to
therapy. However, if overall drug development is pursued in the
larger population, the effectiveness and risk-benefit will be
evaluated in that population, and approval decisions will be based
on the overall database.
Much of the
concern about FDA actions in this area is based on the perception
that pharmacogenomic testing is likely to give definitive answers
about the probability of safety and effectiveness in
subpopulations. Such specificity may occur occasionally (e.g.,
where a product is designed to inhibit a specific molecular
target), and in such cases, rapid development of a diagnostic test
is highly encouraged. However, this is unlikely to be the
ordinary case. In most instances, a genotype or particular gene
expression profile is likely to be one of a number of factors that
affects the probability of an adverse event or a favorable
response. For this reason, pharmacogenomic biomarkers can
ordinarily be handled like other non-genomic predictive markers in
the clinical arena.
Application number ________ (leave blank if
this is the first submission for a stand-alone VGDS)