| NCBI Home | |
| Introduction and Overview |
|
The NCBI Entrez Genome Project database is intended to be a searchable collection of complete and incomplete (in-progress) large-scale sequencing, assembly, annotation, and mapping projects for cellular organisms. The database is organized into organism-specific overviews that function as portals from which all projects in the database pertaining to that organism can be browsed and retrieved (see Figure 1). For example, you will find all information regarding the human genome project on this project page. (Although the Entrez Genome Project database does not include organellar-, phage-, plasmid-, or viral-only genome sequencing projects, data from those projects are submitted to GenBank and are available in the Entrez Nucleotide and Entrez Genome databases. Sequences such as the human mitochondrial sequence will be attached to the human genome project. There is also a special set of resources at NCBI dedicated to Viral Genomes.)
Currently this database only consists of projects that have submitted data to NCBI, intend to submit data, or have received public funding.
Genomic projects will be completely searchable in Entrez. Specific query and search terms can be found here. Projects can also be retrieved by project ID if necessary.
|
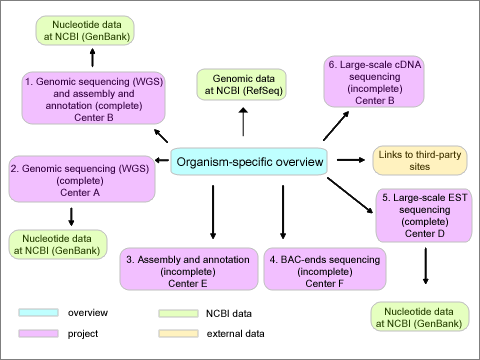
Figure 1. Schematic diagram of a generic eukaryotic genome project. The main overview page (blue) shows links to all subprojects (purple), numbered 1 through 6. Various sequencing centers are associated with each subproject (A through F). These various centers could actually be consortiums consisting of multiple centers. A given center could produce more than one type of project, and data for a given project type could be generated from multiple independent centers. Some of the projects are complete with associated data (green) present in various forms in different Entrez databases at NCBI, while other projects are in progress with no publicly available data at NCBI. A project could be converted over time from containing preliminary data (ex. WGS) to one where a complete data set is present. RefSeq genomic data is associated with the overview project. Links to third-party sites (orange) which contain information of interest regarding the organism are provided. |
|
|
Prokaryotic projects are organized differently. Since the typical sequencing project for microbes derives all genomic data from a single sequencing center, each microbe for which we have genomic data is considered a single project. If there is more than one strain for a given organism, then links to each are displayed from any of the strains. For example there are currently 8 Staphylococcus aureus strains that have been sequenced including a laboratory strain, and links to each one are displayed in every project.
Comparison.
Entrez Genome Project is a companion database to Entrez Genome. Sequence data is stored in Entrez Genome (as
complete chromosomes, plasmids, organelles, and viruses) and Entrez Nucleotide (as chromosome or genomic fragments
such as contigs). However, Entrez Genome does not collect all data for a given
organism. The Genome Project database, on the other hand, provides an umbrella view of the status of each genome project,
links to project data in the other Entrez databases, and links to a variety of other NCBI and external resources
associated with a given genome project. Sequences associated with a given organism can also be retrieved in the taxonomy browser. However, no
distinction is made between GenBank (non-curated) and RefSeq (curated) sequences. There is also no distinction
based on which sequencing center submitted the data. Entrez Genome Project also lists projects that are in progress
or for which NCBI has not yet received any data. See Table 1 for a comparison of all 3 databases.
| Entrez Database | Organism-specific
sequences |
Project-specific
sequences |
Submitter-specific
sequences |
Complete and
in progress |
GenBank and
RefSeq sequences |
| Genome | Yes | No | Yes | No | Separated |
| Taxonomy | Yes | No | No | No | Together |
| Genome Project | Yes | Yes | Yes | Yes | Separated |
New genome sequencing projects can be registered through the Genome project submission form.
Information on the submission of projects can be found here.
More information about the submission of data from complete genomes is provided in the Resource Guide section on Submission of complete genomes.
If you have any additional questions, then please send an email to:
info@ncbi.nlm.nih.gov
| Project Submission Form |
|
The submission form has recently been changed. Some fields have been eliminated to make the form simpler and on a single page, while new fields have been added. Tool tip help messages are available for each field and will appear in the box on the right side of the page. If you have any questions, then please send an email to: genomes@ncbi.nlm.nih.gov
The purpose of the genome project submission page is twofold: to allow a place for genome sequencing centers to register their projects and locus tags as well as a starting point for the submission of genomic sequence data. Submitters need not submit sequencing data at the time they register their project. This section describes each field in the submission form.
Required fields are contained within the top section of the page and optional fields are in the bottom section of the page. Failure to fill out the required fields will prevent the submission from being sent to NCBI and a warning message will inform submitters of any problems that need to be corrected before the submission can be sent.
Please note that sequencing projects that consist SOLELY of organellar-, phage-, plasmid-, or viral-only genome sequences do not need to have their locus_tag prefixes registered, and a Genome Project submission form does not need to be filled out. Plasmids that are part of a complete genome (chromosomes and associated plasmids) from an organism will be registered as part of the complete genome sequencing project.
Once you have submitted a project, you will receive a project ID in a return email. Please use this identifier in the subject line of your email in all subsequent information updates and in communications with NCBI staff. NCBI staff will contact you shortly after receiving your submission. Note that genome projects will no longer be kept confidential. Once submitted, they will be accessible and searchable through Entrez Genome Project. This does not mean that the DNA sequence will be publicly available but the information in Genome Project (organization, organism, locus tag prefix, type of project, etc.) will be viewable by the public.
| Required Info |
|
| Project type |
|
The first required field determines the type of project data. Select the appropriate button for projects consisting either of genomic data from a single organism or from projects consisting of metagenomic or environmental genomic data (sequences from multiple organisms). Depending on which button is selected here specific fields in other parts of the submission form will be affected.
| Contact |
|
Please provide a name for the contact person that NCBI staff can send correspondence to. Include their first and last name as well as a valid email address. This last part is essential as we cannot contact a submitter without a valid email address. If the submitter wishes to include additional contacts they can click on the 'Add another contact' link which will open a new set of fields.
| Submitting organization(s) |
|
One of the three submitting organization fields must be filled in. Submitting organizations consist of three types:
Submitting Organization are usually a Sequencing Center, University, or Research Laboratory where the sequence data is generated and analyzed. In certain cases, the Sequencing Center may differ from the Submitting Organization so space is provided to enter both Organizations. If more than one Organization or Sequencing Center was involved, then click on the 'Add another organization' or 'Add another center' links which will open a new set of fields. The third type of submitter is a consortium which is an umbrella organization for a set of collaborations between different organizations. It is expected that there will be only one consortium for a specific sequencing project.
In all cases there is a field provided for a URL for each organization (homepage). The URL is not required for submission of the project. Project-specific URLs are in the optional section of the submission form (see below).
| Organism/project name |
|
Depending on which project type was selected in the project type field, the appearance of this field will differ:
If a single organism project was selected, then this field is for that organism's scientific name (ex: Escherichia coli). If a metagenomic project was selected then the name of that metagenomic project should be entered here (ex: 'Northern Pacific Deep Sea Vent Metagenomic Project') so that it can be distinguished from other metagenomic projects.
Please note that sequencing projects that consist SOLELY of organellar-, phage-, plasmid-, or viral-only genome sequences do not need to have their locus_tag prefixes registered, and a Genome Project submission form does not need to be filled out. Plasmids that are part of a complete genome (chromosomes and associated plasmids) from an organism will be registered as part of the complete genome sequencing project.
The organism name is of tantamount importance for the submission of a genome project as many functions at NCBI are linked via taxonomic listing. Please provide the most complete scientific organism name possible. If the name is modified by a subspecies, serotype, or other qualifier, then include that in the organism name.
| Strain/isolate/breed |
|
The strain/isolate/breed name is important for differentiating two genome projects that come from the same organism. Due to the ever-increasing numbers of genome projects, this field is now required. Strain names for bacteria (ex. K-12 or ATCC 10987) should be entered as they appear in the literature. The same is true for breeds or isolates or any other marker that distinguishes this organism from another. Note that if a metagenomic project was selected, then a strain/isolate/breed identifier is not required and the field will not appear in the form.
| Locus tags |
|
Submitters of projects can now register locus_tag prefixes for their prokaryotic (and eukaryotic) genome sequencing projects.
What is a locus_tag? In the era of genome sequencing, it became necessary to uniquely identify every gene in a genome, especially when a gene name had not yet been assigned, or when the function of that gene had not been identified. To that end the locus_tag was invented as a means to uniquely identify every single gene in a given genome. The rapid increase in genome sequencing projects has led to some locus_tags being used in two or more different genomes, which has led to confusion when searching the sequence databases or when reading the scientific literature. Therefore, it has become necessary to ensure that locus_tags are now unique across all genomes, and so we have provided a way to search existing locus_tag prefixes, as well as a way to register them during submission of a project.
Existing locus_tags can be searched here.
Locus_tags must now be registered. To assign your own locus_tag prefix, check the availability in the webpage above. If the prefix you wish to register is available, then it can be added to your submission form. If that prefix already exists, then an error message will pop up and a different prefix will have to be used. If the prefix has not been registered previously, then a popup window will inform the submitter that that prefix is available. At that point the prefix can be registered along with the sequencing project. Note that if the locus_tag prefix field is left blank, then a prefix will be automatically assigned from the list of available prefixes.
More information on the joint proposal from the American Society for Microbiology and the National Center for Biotechnology Information is available in this document.
Information on feature table qualifiers can be found here, and locus_tags in the list of qualifiers.
| DNA source |
|
It has been brought to our attention that it can be extremely difficult to find the source DNA/bacteria used in sequencing projects
and in some cases that DNA is no longer available.
Sometimes the submitting centers are not the ones who have the source DNA or bacterial strain. Therefore, providing
this information will enable access to the DNA for all interested parties.
If the bacterial strain has been submitted to a culture collection, then enter the name of the cutlture collection
where it is stored (ex: 'Bacterial strain ATCC 10987 available from ATCC). If the source is not bacterial or
the DNA is stored somewhere other than a culture collection, then enter the name and email of a contact person (ex: Source DNA available from Dr. John
Doe (jdoe@dnares.edu) at the University of Research).
| Optional fields |
|
There are far fewer optional fields than were present in the previous submission form. None of these fields are required for submission but they do provide additional information for the scientific community.
| Replicons |
|
The replicon fields capture the chromosome/plasmids names for each complete DNA molecule present in this specific Genome Project. Enter the name (ie. chromosome I), size (in megabases) and type (chromosome/organelle/plasmid) for each replicon (the default is chromosome). Two fields are present to start with and more can be added by clicking on the 'Add more' link. If a replicon needs to be deleted, click on the 'remove' icon to the right of each row.
| Method |
|
The method drop-down menu provides a number of method-types for genome sequencing projects. Four methods are available:
| Sequencing depth |
|
Enter the average sequencing depth for the genome sequencing project (ex: 8X).
| URL |
|
Many projects have their own specific web addresses where information and data about the genome sequencing project are stored and accessible to the public. If the submitting organization maintains a specific project page, then enter the URL here. If data is available on a separate webpage, then enter the URL in the Data URL field. These two URL fields (project-specific) will differ from the one present in the required section (organization-specific).
| Estimated genome size |
|
Enter the estimated genome size (in megabases) for the genome sequencing project. Published bacterial sequencing projects will have a genome size calculated based on the deposition of sequence data. Eukaryotic projects will use the estimated/curated genome size due to their complexity.
| Project description |
|
Enter a description of the organism in the box provided. This should include the genetic, medical and historical relevance of the organism and why it is being sequenced. If a metagenomic project was selected, then explain the significance of the particular environment that is being investigated.
| Properties of Eukaryotic Genome Sequencing Projects Table |
|
The Properties of Eukaryotic Genome Sequencing Projects page displays selected properties of eukaryotic genome sequencing projects in a sortable and filterable table. The rows of this table represent projects, and the columns contain the properties. This table includes properties of genome sequencing projects only, and it reflects the information contained in the Genome Project database; it does not include properties of assembly, annotation, map, EST, or other non-genome-sequencing projects. The properties, which are hyperlinked to relevant resources when available, are organized into organism-specific information, sequence-related information, and general links.
Note: Use of the word "property" does not indicate that the property names can be used in a property-restricted query (e.g., GB[property]) to retrieve the corresponding data.
| Properties: Organism Information |
|
Properties under the Organism Information heading pertain to the sequenced organism. They consist of Organism, Group, Subgroup, TaxID, Genome Size, and # Chr.
Organism
The Organism property is the species and strain (when applicable) of the sequenced organism. It is
hyperlinked to the Genome Project overview page for that organism; from there, the specific genome sequencing project page can be accessed.
Group
The Group property is the common name of the general organismal group to which the sequenced
organism belongs. Possible values for this property are Animals, Fungi, Plants, and Protists.
Subgroup
The Subgroup property categorizes the sequenced organism within a Group. For example, organisms in
the Animals Group may belong to the Mammals, Birds, Fishes, Flatworms, Insects, Reptiles, Roundworms, or Other Animals (none of the just named) Subgroup.
TaxID
The TaxID property is the taxonomy ID assigned to the sequenced organism in the NCBI Taxonomy
database. It is hyperlinked to the NCBI Taxonomy Browser record or node for that taxonomy ID.
Genome Size
The Genome Size property is the haploid genome size (in megabases [Mb]) of the sequenced organism, when
known. It may be either the calculated size, based on sequence, or the estimated size, based on the literature or other resources.
# Chr (Number of Chromosomes)
The # Chr property is the haploid number of chromosomes that the sequenced organism has, when known.
| Properties: Sequence Information |
|
Properties under the Sequence Information heading pertain to the specific genome sequencing project. They consist of Status, Method, Depth, Release Date, Center/Consortium, GB, and PM.
Status
The Status property refers to the current stage of the sequencing project. Possible values for this property
are Complete, which typically means that each chromosome is represented by a single scaffold of very high sequence quality; Assembly, which typically means that scaffolds have been constructed that are not yet at the chromosome level and/or are of draft sequence quality; and In Progress, which indicates that either the sequencing project is at the pre-assembly stage or the assembled/completed sequences have not yet been submitted to GenBank/EMBL/DDBJ.
Method
The Method property refers to the sequencing method(s) used to sequence the organism's genome.
Possible values include Clone-based sequencing, Whole Genome Shotgun (WGS) sequencing, WGS and
Clone-based sequencing, and Array Re-sequencing.
Depth
The Depth property is the average-fold genome coverage achieved in the sequencing project.
Release Date
The Release Date property reflects either the earliest date that sequence generated in the sequencing project
became public in GenBank/EMBL/DDBJ or the date of a publication, press release, or other announcement indicating
that the sequence has been released. The Release Date value is approximate; it has the format month/day/year.
Center/Consortium
The Center/Consortium property indicates the sequencing center(s) and/or consortium(s) that are
associated with the genome sequencing project. If there is only one center/consortium, its name is displayed and hyperlinked to the center's/consortium's Web site, when available. If there are two or more centers and/or consortiums, the name of only one is displayed and "[more]" is appended; clicking on the name displays a popup window containing a listing of the individual centers/consortiums, each of which is hyperlinked to its corresponding Web site, when available. If the members of a consortium are specified in the Genome Project database record for the project, they will appear in the popup list.
GB (GenBank Accessions)
The GB (GenBank Accessions) property provides access to the GenBank/EMBL/DDBJ record(s) of the
sequence generated in the genome sequencing project and deposited in GenBank/EMBL/DDBJ. It is hyperlinked to the Summary view of the project's GenBank/EMBL/DDBJ accessions (including WGS project accessions) in Entrez Nucleotide.
PM (PubMed)
The PM (PubMed) property provides access to project-specific publications. It is hyperlinked to the
Summary view of the project's publications in Entrez PubMed.
| Properties: Links |
|
Properties under the Links heading indicate availability of, and provide access to, data and analysis tools that are specific to the organism that was sequenced; they are not project-specific. These properties consist of R (RefSeq Accessions), G (Entrez Gene), T (Trace Archive), B (BLAST), M (Map Viewer), and F (FTP Sites).
R (RefSeq Accessions)
The R (RefSeq Accessions) property provides access to genomic RefSeq record(s) representing whole chromosomes and intermediate assemblies for the sequenced organism, when available. It is hyperlinked to the Summary view of the organism's RefSeq accessions (NC_ , NT_, and/or NW_) in Entrez Nucleotide.
G (Entrez Gene)
The G (Entrez Gene) property provides access to Entrez Gene records for the sequenced organism; it is available only for those organisms represented in Entrez Gene. This property is hyperlinked to the summary view of the organism's gene records in Entrez Gene.
T (Trace Archive)
The T (Trace Archive) property provides access to trace sequences that are derived from the sequenced organism and have been deposited in the Trace Archive. It is hyperlinked to the organism's trace sequences in the Trace Archive.
B (BLAST)
The B (BLAST) property provides access to a BLAST page that performs organism-specific, group-specific (such as fungi-specific), or eukaryotic-specific BLAST analyses. It is hyperlinked to the most specific BLAST page available for the sequenced organism.
M (Map Viewer)
The M (Map Viewer) property provides access to Map Viewer maps for the sequenced organism. It is hyperlinked to the organism's Map Viewer genome view page.
F (FTP Sites)
The F (FTP Sites) property provides access to RefSeq and/or Map Viewer FTP files for the sequenced organism, depending on availability. An "F" indicates that either RefSeq FTP files or Map Viewer FTP files are available; it is hyperlinked to the corresponding FTP site at the level of the most specific FTP files available for the organism. An "F+" indicates that both RefSeq and Map Viewer FTP files are available; clicking on it displays a popup window containing a listing of the two corresponding FTP sites, each of which is hyperlinked to that FTP site at the level of the most specific FTP files available for the organism.
| Sorting |
|
To facilitate the viewing of information in the Properties of Eukaryotic Genome Sequencing Projects table, the projects (rows) can be sorted based on a single property or on multiple properties. If GB, PM, R, G, T, B, M, or F are selected as a sorting property, the sort is based on the presence or absence of that property in a row. By default, the projects in the table are sorted by Organism only, in ascending order (i.e., the rows are arranged based on the alphabetization of the Organism field from A to Z). Sorting of the properties (columns) is not available.
Sorting by a single property
To sort using a single property, select the property:
Sorting by multiple properties
The projects can be sorted based on multiple properties by first sorting using a single property, and then performing sequential sorts with each additional property individually. Each successive sort will maintain the orderings achieved in the previous sorts.
To sort using multiple properties, first select a property for the primary sort:
| Filtering |
|
A subset of projects (rows) in the Properties of Eukaryotic Genome Sequencing Projects table can be selected for display by filtering based on the Group/Subgroup, Status, and/or Method properties. Selection of a subset of properties (columns) for display is not available.
To filter, use the pull-down filtering menus located above the table:
To clear filtering:
| Saving |
|
The Properties of Eukaryotic Genome Sequencing Projects table can be downloaded and saved as a tab-delimited text file.
To save:| Prokaryotic Genome Project Tables |
|
The genome project tables are available from the main page.
The prokaryotic table contains three tabs, one for complete genomes, one for in-progress genomes, and a unique tab that presents organism information derived from the attribute table. Each table provides filtering and sorting abilities. Every tab provides filtering abilities by organism group, while the Organism info and Genomes in progress tabs provide additional filters based on sequence status. For the Organism info tab, projects can be filtered based on "all", "complete", "assembly", or "no sequence or blast only" projects. The Genomes in progress tab provides filtering based on "all", "blast only", "WGS assembly", or "no sequence available" projects. The Complete genomes tab only provides complete genomes that are publicly available. The columns with green titles can be sorted, and multiple column sorting is present. Sorting can be in ascending or descending order. If any filters are set, they can be reset to the default by clicking on the clear filter button on the grey toolbar. The information in the table can also be saved as a text document by clicking on the "save" button on the same toolbar. Table rows are color-coded in the Organism info and Genomes in progress tabs, while in the Complete genomes tab they are colored light green and grey to increase readability. For Genomes in progress, the colors are: light green (BLAST only), light blue (WGS assembly), and grey (no sequence available). For Organism info, the colors are: dark green (complete), light blue (assembly), and light green (no sequence or BLAST only).
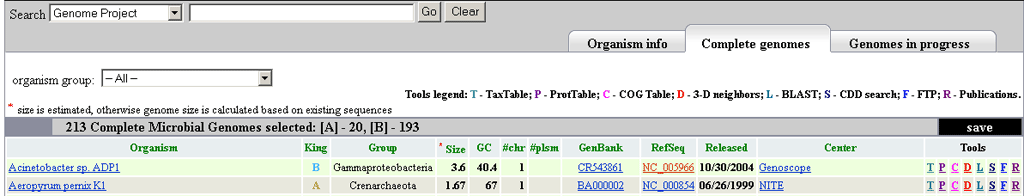
The complete tab shows all complete genomes present at NCBI (213 when the image was captured). The columns are: Organism, Kingdom, Organism Group, Genome Size (estimated if with an asterisk, otherwise the total genome including all chromosomes and plasmids), GC content (total GC content including all chromosomes and plasmids), number of chromosomes, number of plasmids, GenBank and RefSeq Accessions for the largest chromosome, the release data, the sequencing center, and the tools column. The organism links go directly to the Genome Project display, while the GenBank and RefSeq columns are linked to Entrez Nucleotide and Entrez Genome, respectively. The Center column links directly to the sequencing center's webpage for that particular sequencing project (if one exists), otherwise the link goes to the sequencing center's mainpage. The tools column provides links to the TaxTable, ProtTable, COG Table, 3D neighbors, genomic BLAST, CDD search, the RefSeq FTP directory for a given genome, and a link showing publciations associated with a given genome and/or project.
The Genomes in progress tab provides filtering for both organism group and sequence status. The columns are: Organism, Kingdom, Organism Group, number of Contigs, Genome Size (estimated if with an asterisk, otherwise the total genome including all chromosomes and plasmids), GC content (total GC content including all chromosomes and plasmids), RefSeq Accession, Genomic BLAST, and sequencing center. The organism links go directly to the Genome Project display, while the RefSeq Accession goes to Entrez Genome. The Center column links directly to the sequencing center's webpage for that particular sequencing project (if one exists), otherwise the link goes to the sequencing center's mainpage. There are no tools provided for in progress genomes. In the image shown, the In progress tab has been filtered for the Deltaproteobacterium group, and the number of contigs has been sorted in descending order.

The Organism info tab provides filtering for both organism group and sequence status. The columns are: Organism, Kingdom, Genome Size (estimated if with an asterisk, otherwise the total genome including all chromosomes and plasmids), GC content (total GC content including all chromosomes and plasmids), Gram stain, Shape, Arrangement, Endospores, Motility, Salinity, Oxygen Requirements, Habitat, Temperature Range, Pathogenic in (host), and Disease. The organism attibutes are derived from the individual attributes present in each genome project. Those attributes are explained below. The organism links go directly to the Genome Project display,

| Prokaryotic Attributes Table |
|
The prokaryotic attribute table describes various physiological features associated with a given microbial organism. It is not intended to be the definitive guide to bacterial differentiation. For that, NCBI suggests consultation of a manual such as Bergey's Manual of Determinative Bacteriology, or Bergey's Manual of Systematic Bacteriology. This table is merely intended to be a general guide to prokaryotic organisms for which genomic information is present at NCBI.
Not all fields are completed for all bacteria. If there is missing information for which
you wish to add to a given organism, then please supply a publication citing the information
and send an email to:
This table is present on each individual genome project display page. The information present in these tables are also collected and shown on the organism info@ncbi.nlm.nih.gov
| Cellular features |
|
Cellular features describe the general characteristics of a given bacterium. There are 5 categories including gram stain, shape, cellular arrangement, presence or absence of endospores, and motility.
| Gram stain |
|
The gram stain is one of the oldest methods used to differentiate bacterial cells and was developed by Christian Gram in 1884, for which the procedure is named. The process is based on a staining procedure that results in either a positive or negative stain being observed. Gram positive bacteria are ones which have a cytoplasmic membrane surrounded by a peptidoglycan layer. Gram negative bacteria have an additional outer membrane outside of the cytoplasmic membrane and peptidoglycan layer. This field is a controlled vocabulary of either "positive", "negative", or null, if no gram stain has been performed on the organism or no information about the staining results are known.
| Shape |
|
The shape field consists of uncontrolled vocabulary that describes the basic cellular shape of the organism. Examples include, coccoid (spherical), rod-shaped, or spiral-shaped.
| Arrangement |
|
Arrangement describes whether the organism exists solitarily, or whether it can be found in groups of 2 or more cells. The vocabulary for this field is uncontrolled. For example certain bacteria, such as the cyanobacteria, form filamentous structures consisting of undifferentiated (phototrophic) cells punctuated by a differentiated (nitrogen-fixing) cell.
| Endospores |
|
This field consists of a controlled vocabulary of "yes" if the organism produces endospores, no if it does not, or null if unknown. Many important organisms produce endospores during times of environmental stress, allowing them to persist until more favorable conditions develop. For example, the anthrax bacterium, Bacillus anthracis, produces endospores that are infective.
| Motility |
|
The motility field describes whether the organism is known to be motile or not. It consists of a controlled vocabulary. "Yes" means the organism has been found to be motile, while null means it has not, as of yet, been shown to be motile. Motility may be a result of the production of extracellular appendages such as polar flagella, peritrichous flagella, or other appendages such as type IV pili that are involved in twitching motility.
| Environment |
|
The set of environmental fields describe the basic environment in which the organism prefers or has been found to live in. The fields include salinity, oxygen requirements, and habitat.
| Salinity |
|
Salinity is a controlled vocabulary field that describes the salinity requirements of the bacterium (percentage of salt as sodium chloride equivalent in the growth medium). The following 4 categories apply:
| Oxygen Req. |
|
Oxygen requirements describes the ability of the organism to live at various levels of oxygen. The following 5 controlled categories apply:
| Habitat |
|
This field describes the basic environments in which the organism is found, however, it is not intended to reflect ALL possible environments. The 6 controlled categories are:
| Temperature |
|
These two fields describe the optimal and range of temperatures (in Celsius) the organism grows at.
| Opt. temp. |
|
| Range |
|
Temperature range is a controlled vocabulary field that describes the basic category of temperature range the organism grows at. Organisms that grow at ranges that overlap multiple categories are classified based on which category the majority of their temperature range overlapped with. The following 6 categories apply:
| Pathogenic In |
|
This field describes organisms that this bacterium is pathogenic in. This field does not contain a controlled vocabulary, and it is not expected that all potential hosts will be listed. This field is related to the disease field below.
| Disease |
|
This field provides the name of the disease causes by a pathogenic bacterium and is related to the field above. This field does not contain a controlled vocabulary, and common names for diseases are often present, as well as multiple names.
| FAQ |
|
| Understanding Genome Projects |
|
| What is a genome project? |
|
Genome projects are hubs that collate organism-specific genomic data at NCBI into a hub and spoke system. This allows easy access to all of the genomic data, whether it be genomic sequences, sets of ESTs, cDNA libraries, mapping projects, or other genomic information. A more comprehensive explanation can be found in the introduction section. Note that NCBI is currently only listing cellular organisms in the projects database, which does not include viruses.
| I don't understand this table on bacterial projects. What does it mean? |
|
This table describes various physiological attributes for bacterial organisms. A more specific description of each field in the table can be found here.
| Querying and searching |
|
Genome projects is integrated into Entrez. This means that the same search terms you would use in nucleotide, pubmed, or the genes database can be used here to find specific information on a genome project.
For tips on searching, view the querying and searching section of this help document.
| How do I submit a Genome Project? |
|
Genome projects can be registered at NCBI by filling out the submission form. Submitters can either submit and register a genome project or submit a genome project and submit genomic data at the same time. Information on submitting the project can be found on the project instruction page. Information on submitting genomic data can be found on the sequence instructions page.
| How do I submit genomic data? |
|
Genomic data can be submitted through the project page. NCBI would prefer if you registered your project first and followed the instructions on submitting genomic data.
| What do I do if I have important information for an existing project? |
|
If you have data already at NCBI that you would like to link to an existing project, then please send an email to:
info@ncbi.nlm.nih.gov
Include information on the organism you wish to link your data to, the project ID,
the type of data you have, and any identifiers for your specific data set. Examples
include a set of ESTs for a eukaryotic genome project, in which case you would need
to send us the identifiers for the EST sequences, or a microarray project in GEO.
If you have data or information that is on an external website to NCBI, and would like us to provide a link to your site, then send an email to the address above and NCBI staff will provide a link to your website.
| What is a project ID? |
|
Project IDs are unique identifiers assigned to each project. Once a project is submitted, a unique project ID is assigned to it. It is recommended that a submitter should use this project ID in the subject line of any email correspondence with NCBI. This allows easier tracking and faster processing of submissions. The project ID can also be used to search in Entrez Genome Projects.
Project IDs will be assigned to all genomic sequencing projects which are derived from a single organism/metagenomic project from a submitting organization. Project IDs will be used as an umbrella ID that points to all genomic sequencing data and will appear on all sequencing records that are derived from a single sequencing project. This is already being done for the Reference Sequence project at NCBI (example: Human chromosome 22)
Project IDs should not be confused with Accession Numbers. INSD will use project IDs for the registration of locus tags and for the submission of genome sequencing projects.
| How do I use a project ID? |
|
Project IDs should be in the subject line of any email correspondence with NCBI regarding genome projects. The project ID can also be used to search in Entrez for a specific project.
| Querying and Searching |
|
The genome projects database utilizes all of the features of other Entrez databases. You can limit searches, preview/index your search terms, use the history or clipboard, or details, all by using the tab buttons underneath the search box. More general instructions on Entrez querying can be found here
| Limits |
|
The limit button at the bottom left hand corner of the grey search bar allows access to the limits page. From this page searches can be limited to the most relevant parameters. If you wanted to search for only those genome projects that had a particular attribute, then you would select that parameter from the drop down menu and enter the search term associated with that menu selection. For example, if you wanted to construct a search query that would only return complete sequences, then you would select sequencing status from the drop down menu, type "complete" in the search box, and hit the GO button.
After selecting a limit the currently selected field will show up in the yellow bar behind the Field tag. The limits checkbox will also be marked and will remain through subsequent searches. To remove the limits for a particular search, deselect the checkbox.
The following table summarizes the various limits and properties that can be used to refine searches.
| Field name | Definition [including field abbreviations] | Examples |
|---|---|---|
| Accession | Accession number associated with a given genome project. This can include GenBank, DDBJ, and EMBL, or RefSeq Accession Numbers, but not protein Accessions.
[CENTER][SEQUENCINGCENTER] |
Retrieve records containing the accession NC_003197:NC_003197[ACCN] |
| Center | Sequencing center associated with a given project, either as the submitting center, as a collaborator, or as part of a consortium.
[ACCN][CACCESSION] |
Retrieve all projects associated with The Sanger Institute:Sanger[CENTER] |
| Chromosome GI | Chromosome GI is the unique identifier used in Entrez genome to refer to a single molecule, whether it be a
single plasmid, a single chromosome, or whether it refers to a set of contigs that all come from the same chromosome.
[Chromosome_GI][GI][CHRGI] |
Find all genome projects associated with chromosome GI 24 24[CHRGI] |
| Creation date | Date the record was created. Note the format is: YEAR/MONTH/DAY including the forward slashes.
[CDT][CREATEDATE] |
Find all projects created between January 1, 2003 and December 31, 2003: 2003/1/1:2003/12/31[cdt] |
| GC Content | The GC content is calculated from the nucleotides in the chromosomes.
[GC][GCCONTENT][GPLUSC] |
Find all projects where the genome GC content is 50%.
50[GC] |
| Genome Size | Genome size is calculated from the sum of all nucleotides in Entrez.
[GENOMESIZE][GSIZE][SIZE] |
Find all projects where the genome size is between 1 and 5 mega base pairs (Mbp).
1:5[GENOMESIZE] |
| Organism | The organism associated with a project(s).
[ORGN][ONAM][TAXID][TXID][ORGANISM NAME] |
Find all projects associated with the species Staphylococcus aureus
Staphylococcus aureus[ORGN] |
| Property | An attribute of a Gene record based on its content.
[PROP][PROPERTY] |
Find all bacterial genome projects that have a plasmid.
has_plasmid[PROP] |
| Release date | Date the record was released. Note the format is: YEAR/MONTH/DAY including the forward slashes.
[RDT][RELEASEDATE] |
Find all projects released between January 1, 2003 and December 31, 2003: 2004[RDT] |
| Sequencing Status | Can be used to find incomplete, in progress, or complete genome sequencing projects.
[SEQS][SEQSTAT] |
Find all complete fungal genome projects.
fungi[ORGN] AND complete[SEQSTAT] |
| Text | Anything found in the body, text, or description of a project.
[WORD][TEXT][AB][TXT][DESCR] |
Find all eukaryotic projects that contain the word parasite in the description.
eukaryota[ORGN] AND parsite[TEXT] |
| Prokaryotic Attributes | The following fields refer solely to microbial genome projects and are reflected in the prokaryotic
attributes table. More information about that table can be found here. Note that some
fields have a controlled vocabulary.
|
Find all projects representing Gram positive organisms. positive[GRAM] Find all projects that correspond to organisms that are aerobic or facultative. (facultative OR aerobic)[OXYREQ] Find all projects that correspond to organisms that grow between 11 and 45 degrees Celsius. mesophilic[TEMPRG] Find all projects that correspond to pathogens that can infect humans. human[HOST] |
| Preview/Index |
|
The Preview/Index page on any Entrez database is a powerful resource to construct useful queries and to view terms that have been indexed under any field name. The table in the previous section described the fields used in indexing the records and provided some representative queries using those fields. This section will:
| History |
|
Use of History in Entrez Gene is consistent with all other Entrez databases. You may refer to the History section of the Entrez help documentation for more information.
| Clipboard |
|
Use of Clipboard in Entrez Gene is consistent with all other Entrez databases. You may refer to the Clipboard section of the Entrez help documentation for more information.
| Details |
|
Use of Details in Entrez Gene is consistent with all other Entrez databases. You may refer to the Details Button section of the Entrez help documentation for more information.
If you have any additional questions, then please send an email to:
info@ncbi.nlm.nih.gov
Revised May 6, 2005